



Introduction
가수분해(hydrolysis)는 현장에서 발생하는 고분자복합소재의 물성변화를 야기하는 중요한 여러 원인들 중의 하나이다. 물성변화의 근본이 되는 기전(mechanism)에 대한 이해가 충분하다면 소재의 특성이 변화하는 원리를 이해할 수 있으며 이에 따른 변화를 방지 할 수 있는 대책을 세우는데 매우 유용할 것이며, 이를 이용하여 고무나 프라스틱의 신소재 및 신부품 개발을 하는 연구소 및 산업현장의 인력들에게 도움이 될 것이라 확신한다. 본 편에서는 가수분해의 기전 및 측정방법, 실란(silane) 및 아미드(amide)의 가수분해의 예를 예시하였다.
Experimental
가수분해는 수분이 분자사슬에 가해져서 수분의 영향을 받은 분자의 사슬이 분해를 일으킨다는 것을 의미한다. 복합소재 내에 존재하는 수분은 가수분해(hydrolysis)에 영향을 미친다. Non-glass용기내에서 중성(neutral), 저 이온(low ionic) 상태의 고순도의 수분과 접촉하고 있는 상태에서는 실란(silane)은 수주 혹은, 달 동안 안정한 상태를 유지한다, 하지만, 수돗물과 접촉을 할 경우 알콕시실란(alkoxysilane)의 가수분해는 수시간 내에 상당히 진행된다.2 알콕시실란의 가수분해에 적용되는 촉매는 축합반응의 진행에도 적용되며 수상(water-borne) 실란은 위의 대조표준 실란보다 유리 표면과의 접착을 향상시킨다.2 이는 수분(moisture) 내에 존재하는 촉매가 실리카(silica)표면의 실란결합을 현저히 증가시킨다는 것을 의미한다. 1차반응은 수분의 존재 하에 실란화(silanization)반응의 증가, 2차반응은 더 많은 수분함량에 따라 증가한다.3-5 실리카표면의 수분은 실리카와 커플링(coupling) 제와의 반응을 선호한다, 따라서 입자간의 반응을 감소시킨다.4
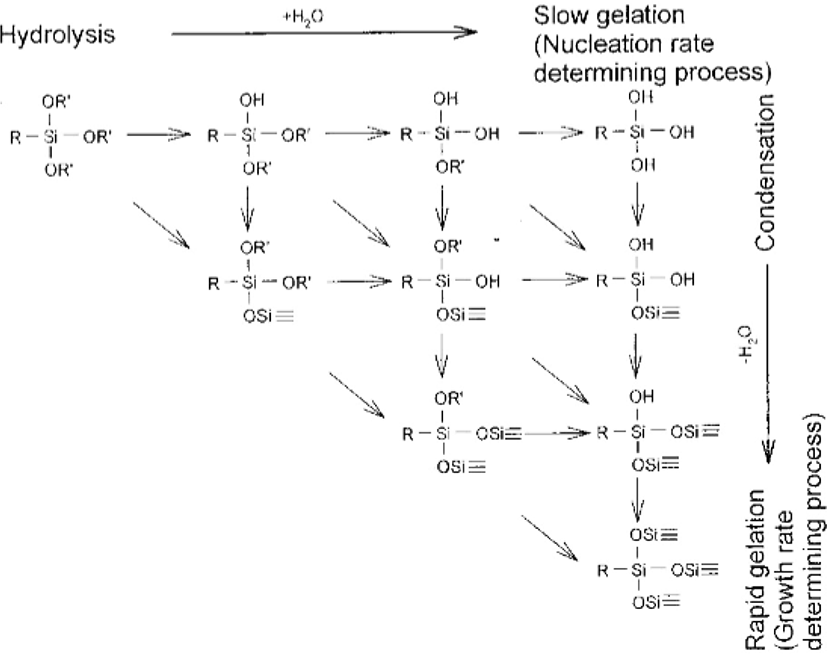
기존의 hydrolysis반응과 축합반응 mechanism에 대하여 다시 정리하였다.6Figure 1은 알콕시실란의 염(base-)과 산(acid-) 촉매작용에 의한 가수분해 기전을 보여준다.
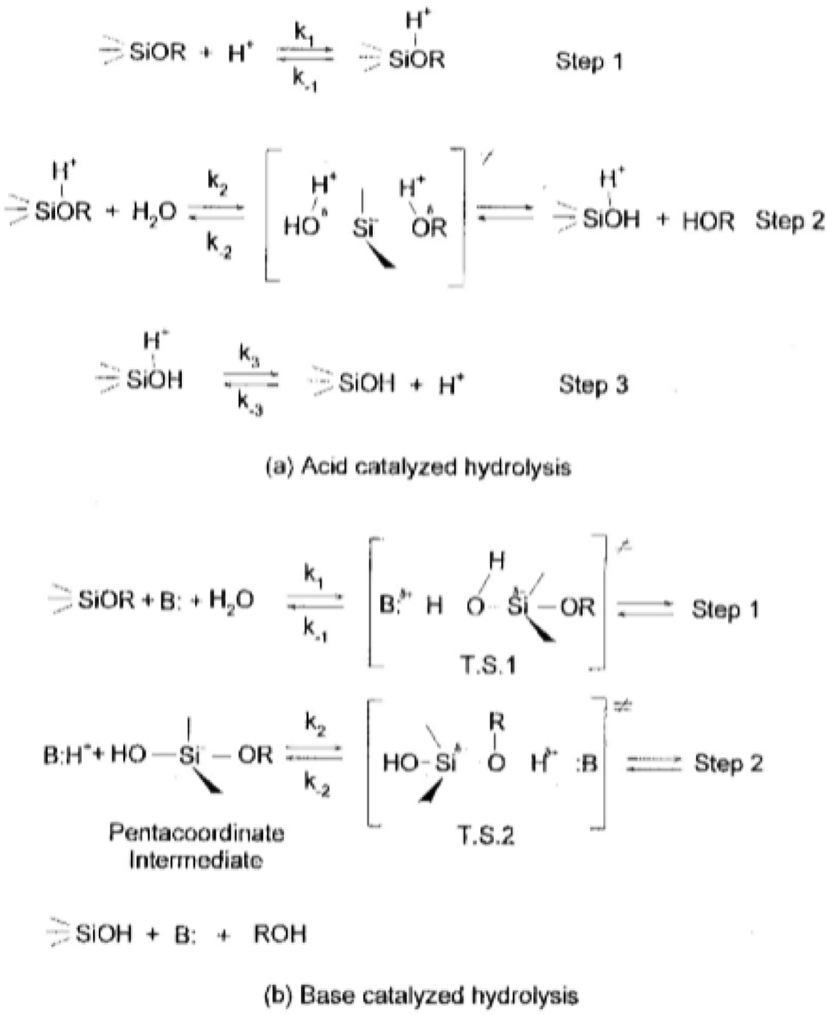
반응이 진행됨에 따라 반응배위(reaction coordinate)는 반응체(reactant)에서 부터 전이상태(transition state)까지 극성 파라미터(polar parameter)값(ρ*)과 입체 파라미터(steric parameter) 값(s)이 구조내 변화와 전하(charge)분배를 정성적으로 보여주고 있다.10
Figure 1(a)는 산촉매작용 가수분해 기전을 보여준다. 실리콘원자 주위에 5개의 치환기들이 SN2-Si의 형태로 존재한다, 접근하는 친핵체와 떠나는 잔여집단(outgoing leaving group)들이 실리콘원자와 하이브리드화(hybridized) 하여 친핵제와 잔여집단간 sp3d 하이브리드화된 5배위 중간생성물(penta- coordinate intermediate) 상태를 형성한다. ρ*와 s 값들이 작다는 의미는 Figure 1(a)에서 보여주었듯이 반응 기전은 기질(substrate)의 평형상태에서의 신속한 양성자화(equilibrium protonation) 후에 수분분자가 잔여집단(leaving group)과 양-분자(bimolecular) 되면서 SN2-type 치환(displacement)을 하는 것이다.7,8
염촉매작용 가수분해 기전이 Figure 1(b)에 예시되었다. 5배위 중간생성물과 함께 양-분자 친핵 치환반응 (SN2**-Si or SN2*-Si)을 한다; SN2**-Si는 5배위 중간생성물 형성 속도결정(k1 < k2), SN2*-Si는 분해(break-down)속도결정(k1 > k2)을 의미한다.7,9ρ*값이 크다는 의미는 전이상태(transition state) 가 실리콘원자에 붙어있는 전자흡수그룹(electron withdrawing group)에 의해 반응물(reactants)보다 더 안정되어있다는 것을 의미한다. 제시된 기전에서는 전이상태(T.S.1 혹은 T.S.2)에서 실리콘원자에서의 음전하(negative charge)가 상당히 높다. 입체파라미터값(s)이 크다는 것은 전이상태가 반응물 보다 입체적으로 더욱 혼잡하다는 것을 의미한다.11 sp3 하이브리드화된 실리콘은 sp3d와 같은 전이상태를 형성하면서 재하이브리드화 되면서 도입되는 하이드록사이드 음이온(hydroxide anion)과 결합을 형성하게 된다. 실리콘표면에 단단히 고정된 음하전밀도(negative charge density)와 친핵 산소간에 단단히 연결된 전이상태를 이룬다.12
Figure 2는 tialkoxysilane의 pH에 따른 hydrolysis mechanism을 나타낸다.13 Allen모델14과 Kay & Assink모델15에 따르면, 염 촉매화 가수분해반응은 알콕시그룹(-(OR)3)이 -(OSi)3 그룹(하단좌측)으로 치환하면 축합반응, 알콕시그룹(-(OR)3)이 하이드록시그룹(-(OH)3)(하단우측)으로 치환하면 이를 가수분해라 부른다. 산 가수분해는 염기 가수분해보다 현저히 크고 다른 카본-결합 치환체들로부터 최소한으로 영향을 받는다. Moore16가 예시한 Osterholtz & Pohl17의 결과는 가수분해가 제일 늦게 일어날 때는 neutral pH이며 pH단위가 1pH씩 변화할 때마다 10배씩 변화한다. 축합반응 역시 pH에 의해 영향을 받는데 반응최소 pH는 4 이다. 알코올의 가수분해반응은 가역적(reverse) 이며 용액을 장시간 동안 안정화시킨다.
자동차의 경량화소재로 잘 알려진 플라스틱(plastic) 소재는 엔지니어링플라스틱(engineering plastic (EnPla))이다. 최근 전기자동차의 등장으로 인해 차량의 NVH성능의 향상이 요구되고 있고 이를 만족하는 소재는 비 금소재속인 플라스틱 소재의 사용이 더욱 확대되고 있다. 이 전에 CAFE (corporate average fuel economy) 규정에 의한 연비증가 및 CO2 절감을 만족하기 위하여 자동차 경량화 부품의 주 소재로서의 적용을 해야 하는 부분도 있기 때문이다.19,20
재활용이 가능한 대표적인 엔지니어링 플라스틱(EnPla) 소재로써 아미드(-CONH-)결합에 의하여 단량체가 연결된 중합체인 폴리아미드(polyamide; PA)는 aliphatic 폴리아미드의 한 종류로 Carothers와 Berchet에 의해 1935년에 최초로 개발되었다.21 PA66는 높은 결정화도에 의한 우수한 기계적 강도와 100~150°C 환경에서 연속사용이 가능한 내열성, 뛰어난 내마모성 등의 장점을 가지고 있는 소재이며 PA66에 대한 연구는22-32 활발히 이루어져 왔다. 다른 구조를 갖는 엔지니어링 플라스틱 소재인 폴리아세탈(polyacetal; POM), 폴리카보네이트(polycarbonate; PC), 폴리에틸렌 테레 프탈레이트(polyethylene terephthalate; PET), 변성 폴리페닐렌옥사이 드(polyphenylene oxide; PPO)들도 함께 널리 사용되고 있 다.33-36
현장에서 사용되는 차량의 파워트레인 마운트(PM) 중의 하나인 엔진 마운트(EM) 부품에 요구되는 소재는 고인장 강도의 물성을 필요로 하는 폴리아미드66/유리섬유(PA/GF) 복합소재들이다. 노즐 부품과 같이 수분이나 glycol에 노출되는 부품은 내가수분해(hydrolysis resistance)의 특성이 있는 소재가 요구되며, 엔진룸에서 발생하는 열(150°C) 및 외부 환경온도(−40°C), 충격, 염화칼슘에 의한 부식, 오일이나 워셔액 등에 의한 부식 등에 노출되는 부품에는 그 부품에 특화된 소재가 요구된다.
가수분해는 PA의 최대 약점으로서, PA의 내가수분해 특성을 향상시켜 소재의 물성 저하를 최소화하고 기계적 강도 저하를 최소화시켜서 고성능 자동차의 부품으로 적용하기 위한 가수분해 저항성이 향상된 PA66 복합소재의 개발이 필요한 실정이며 이로 인해 내가수분해성 소재개발에 대한 연구는 최근에 여러 산업체에서 많은 관심을 갖고 있다.33,34,37-42
Py-GC/MS는 복합재료의 고분자 및 첨가제의 정성분석에 널리 사용되며 분자구조의 정보를 얻을 수 있는 분석 방법이다.43 가수분해를 정량적 측정방법으로는 각 조건의 소재 내 PA 분자구조의 변화를 분석하는 방법(pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS)),18,45 알콕시실란이 포함된 복합소재내에서 가수분해 반응 후 소재 내 알콕시실란분자내 반응하지 않은 잔류알콜의 양을 측정(headspace/gas chromatographic technique 사용)하는 방법44 등이 있다.
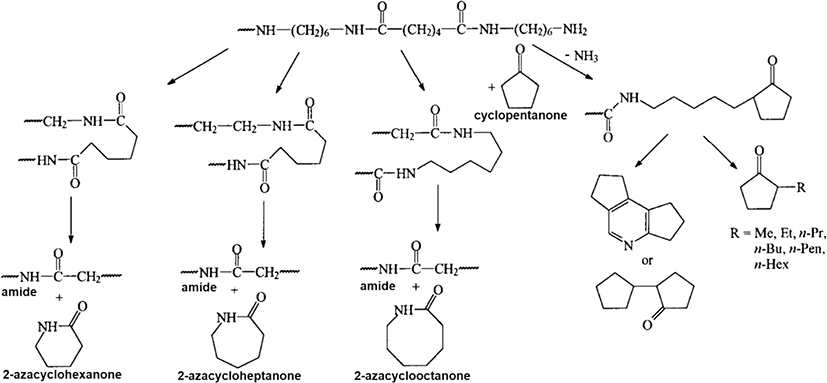
MEG 및 수분의 hydroxyl기가 PA66의 극성 관능기(C=O,-NH)와 상호작용에 의한 결합을 하고 가수분해가 진행되는 것으로 알려져 있다.37 PA66가 열분해되어 생성되는 성분으로는 cyclopentanone, hexamethylenediamine, cyclic monomer 등이 있다.46-50 MacKerron과 Gordon이51 표현한 PA66의 열분해에 대한 기전(mechanism)을 Figure 3에 나타내었으며 PA66의 열분해는 분자사슬의 아디프 산(adipic acid) 분자의 분해부터 시작되어 cyclopentanone, hexamethylenediamine 등이 생성되고, 이 생성된 물질들의 사슬이 절단(scission) 및 재형성(reorganization) 반응을 하여 cyclic monomer가 생성됨은 알려져 있다.42,52
Lee와 Kim은 py-GC/MS을 통해 DTR(D), DuPont(Z), Kopla (K), BASF(A) 사의 PA66/GF(동급) 복합소재들의 열분해 생성물을 정량적으로 확인하고 기계적 물성과 비교하였다.18,42 MEG (mono ethylene glycol)용액에 침지 전후의 크로마토그램을 비교한 결과, cyclopentanone (retention time(r.t.) 5분, ①), 2-N-Hexylaziridine (r.t. 8.6분, ②), 1-Decanol (r.t. 9.5분, ③), 1,6-Hexanediamine (r.t. 11.6분, ④), 3-None-1-ol (r.t. 13.4분, ⑤), Adiponitrile (r.t. 13.5분, ⑤), 1-Methyl-3-formlindole (r.t. 17.8분, ⑥), 2-Azacyclotridecanone (r.t. 21.5~31.3분, ⑦), Hexadecanenitrile (r.t. 30.7분, ⑧) 등의 열분해 생성물이 침지 후 검출량이 공통적으로 증가하는 것을 관찰하였고 Table 1와 같은 결과를 얻었다.
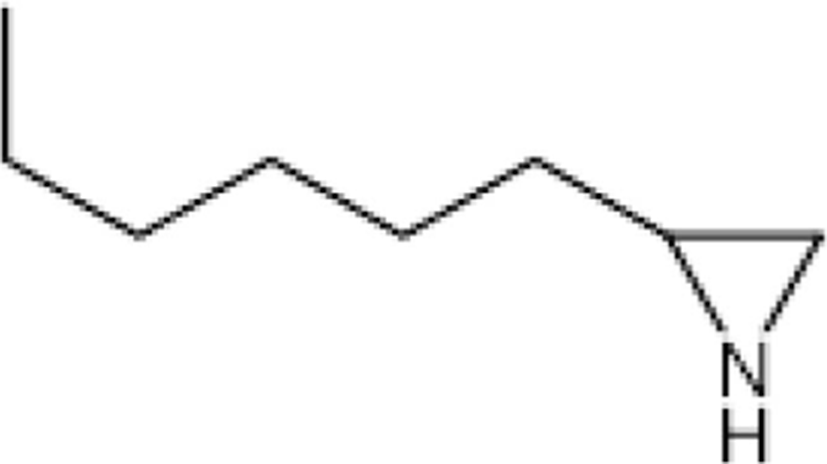

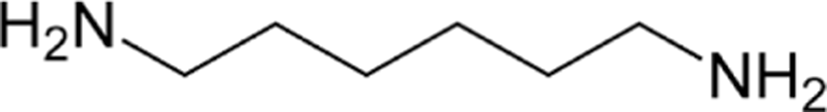
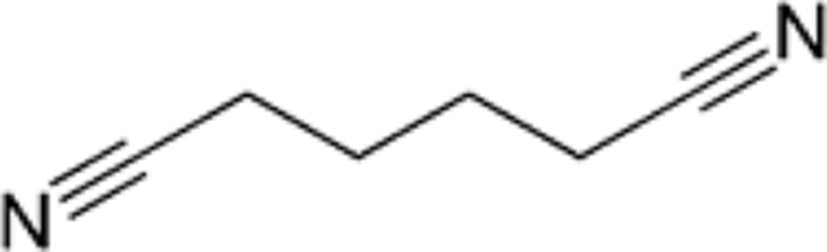
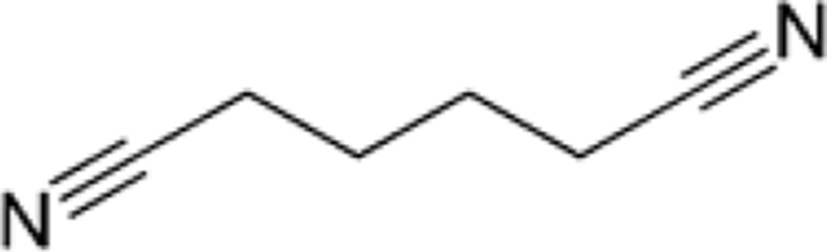
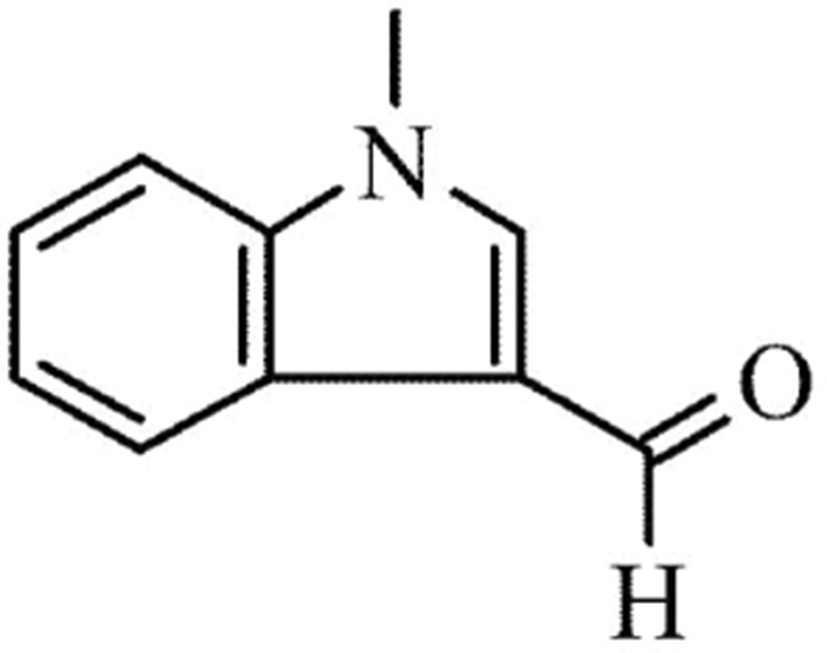
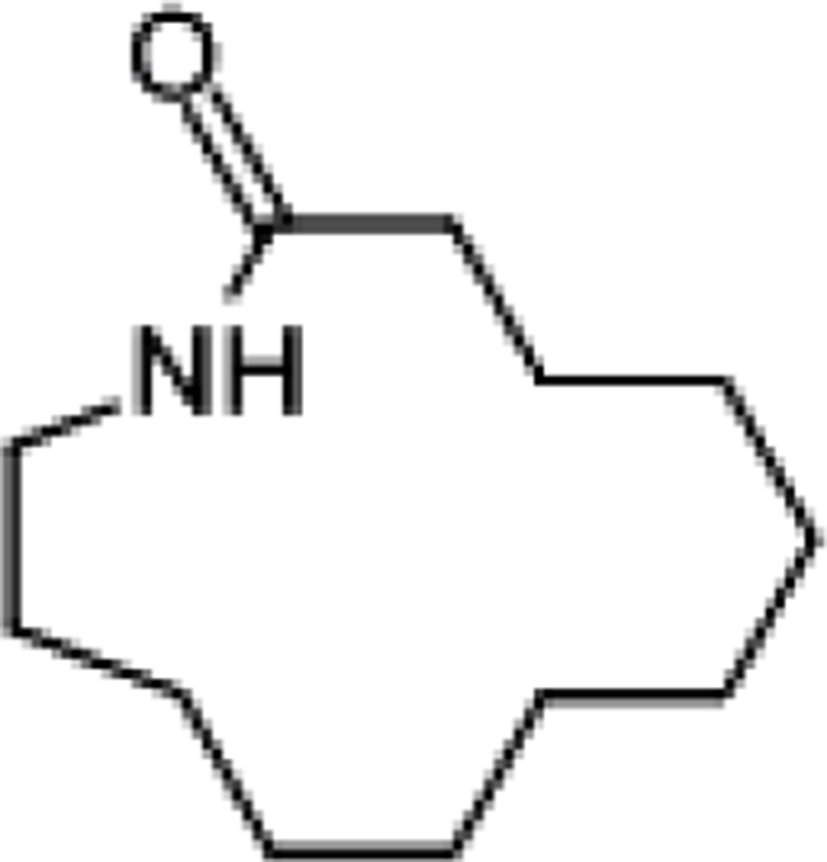

침지 후 PA66 복합소재의 증가한 열분해 물질들의 retention time (r.t.) 및 화학구조는 Table 1에 정리하였다.18,51
이는 함침 시간에 따라 PA66 복합소재 표면에 수분과 MEG의 hydroxy기가 흡착하여 PA66의 사슬의 절단에 관여하여 저분자 및 아디프 산 분자 구조로 분해되기 때문으로 판단되는데, 이는 즉, 가수분해 반응에 의해 MEG 및 수분의 hydroxy기가 PA66의 극성 관능기(C=O, -NH)와 결합함에 따라 carboxylic acid와 alkylamine의 물질이 증가한 것을 의미한다. 침지 전후의 변화된 크로마토그램 특성 피크결과를 Figure 4 에 나타내었다.18
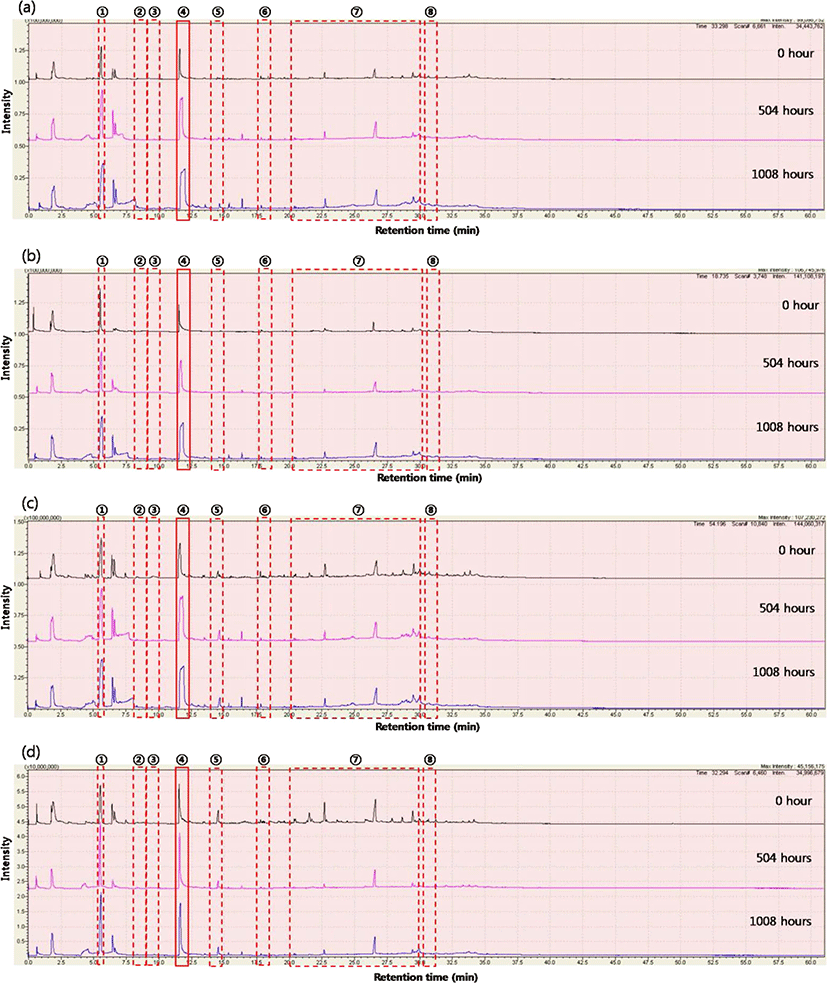
Figure 5는 Figure 4에서 얻어진 질량스펙트럼으로부터 가장 이온강도가 크고, 선택성이 좋은 1,6-Hexanediamine 이온(m/z=30)을 선택하여 정량분석에 이용하였고. GC/MS-selected ion monitoring (SIM)모드로 분석한 결과를 Figure 5에 정리하였는데, 침지 1,008시간 후 각 PA66/GF 복합소재들 (D, Z, K, A)에서 1,6-Hexanediamine 이온의 검출량이 증가함을 확인하였다. 이는 MEG 및 수분의 hydroxy기의 PA66의 사슬과의 반응하여 가수분해가 진행되어 1,6-Hexanediamine 저분자 구조로 분해된 것이기 때문이다. 가수분해로 분해된 고분자는 분자량의 감소에 따른 물성 저하를 보고하였다.53,54 이는 파트2에서 다루겠다.
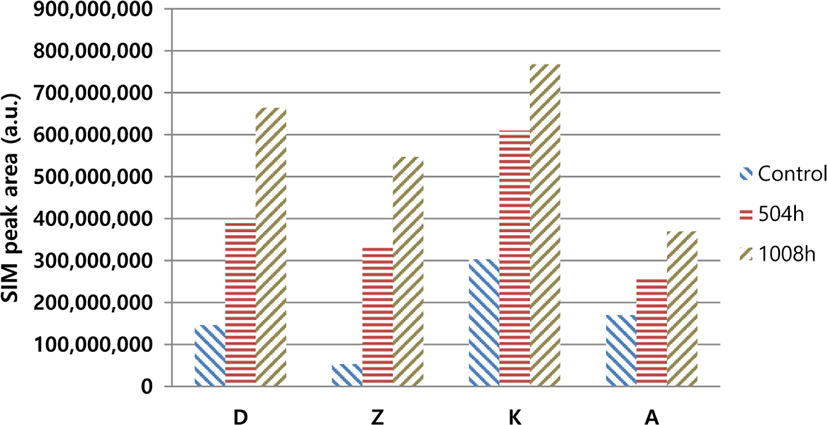
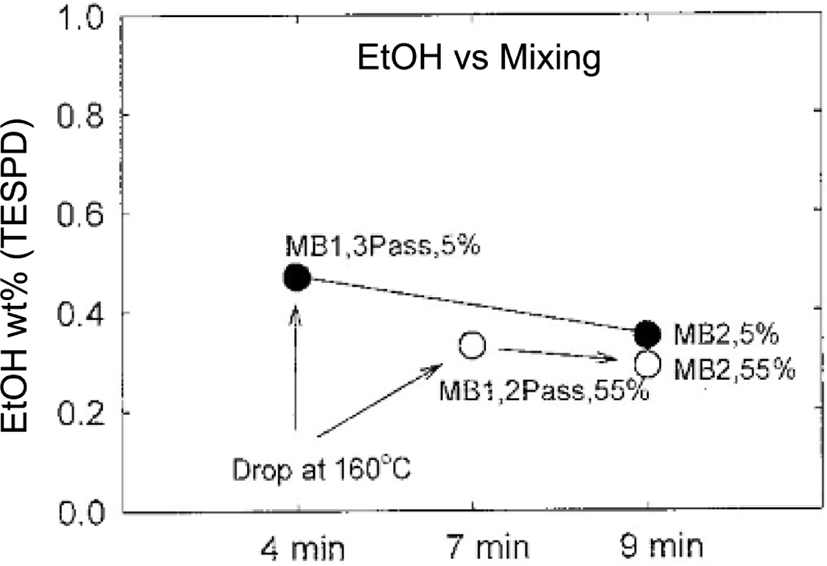
Figure 6는 headspace/gas chromatographic technique으로 측정한 복합소재내 TESPD가 가수분해 후 TESPD내에 반응을 하지 않은 잔류 알코올(alcohol)의 양을 나타낸다.44 믹싱전에 실리카에 수분을 첨가한 소재(open circle)의 알코올 잔유량은 수분을 첨가하지 않은 소재(closed circle)보다 낮았고 또한, 믹싱시간이 증가하면서 두소재의 잔유알콜양이 감소하였다. 이는 수분이 존재할수록, 믹싱시간이 증가할수록 실란과 실리카의 가수분해반응이 증가한다는 것을 의미한다.
양기능성 알콕시실란은(bifunctional alkoxysilane) 직접적 이거나 두 가지 중요단계를 거친다.
Figure 7은 첫째단계로 알콕시실란은 물분자에 의하여 가수분해가 이루어지고 난 후에 수화된(hydrolyzed) 실란은 실리카표면의 hydroxy기와 축합반응을 일으켜 실리카표면에 화학적으로 결합을 하게 된다.44 둘째단계로, 실란의 다른 쪽 끝 황 원소는 고무사슬의 2중결합과 화학적으로 반응을 하게 된다. 이 단계에서 황(S8)고리는 개환(ring open)을 하여 고무사슬의 2중결합과 함께 가류된다. 그리고 나서 실리카와 고무사이에 3차원적 망상조직이 형성된다고 알려져있다.1,56-58,60,61
고분자의 흡수성은 고분자 사슬의 자유 용적(free volume)65,66과 수분과 고분자와의 hydrogen bond와 연계된다.65,66 PA66는 사슬구조 내 산소(O) 및 질소(N) 원자가 물분자와 수소결합으로 반응하여 수분을 흡수하는 특성으로 인해 가수분해에 의하여 PA66 사슬의 절단에 의한 물성이 저하되는 특성을 보이는 것으로 보고되어 있다.67-69 PA 가수분해의 반응 기전(mechanism)을 Figure 8에 표현하였다.70
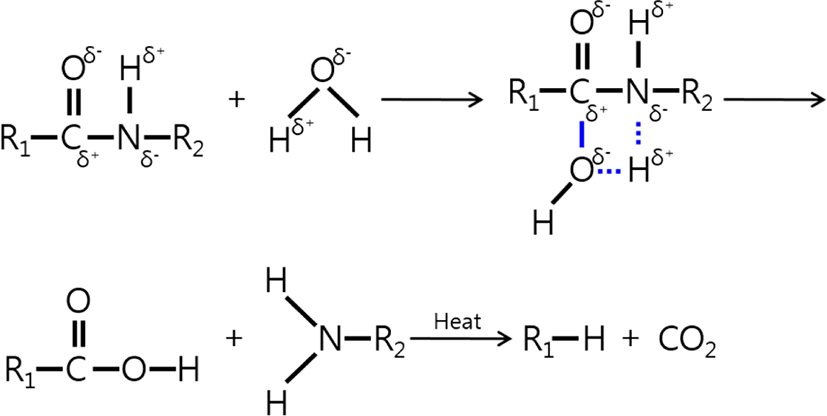
조금 더 자세히 고찰하면, 수분이나 MEG의 극성 관능기인 hydroxyl group은 PA66의 산소, 질소 원자와의 쌍극자 상호작용(dipole-dipole interaction)에 의한 수소 결합을 형성하고, 수분 흡착에 따른 추가 hydroxy기의 증가는 PA 분자 사슬과 GF 표면에서 가소제와 같은 역할을 하여 PA 분자 사슬과 GF간 쌍극자 상호작용을 약화시킨다.28,71-76 수분이 PA 분자 사슬의 부피(volume)와 이동성(mobility)을 증가시켜서 분자들 사이의 얽힘(entanglement)과 결합(bonding)을 감소시키기 때문인 것으로 알려져 있다.70,77,78 이는 인장 신율의 증가로 나타난다.18 가수분해가 진행됨에 따라 복합소재의 인장강도 및 탄성률의 감소, 충격강도, 신장율의 증가. 가수분해된 PA66의 열분해 생성물에 대한 정량평가를 통해 가수분해의 정도를 측정할 수 있다.18,79
MEG 및 수분의 hydroxy기가 PA66의 극성 관능기(C=O, -NH)와 상호작용에 의한 결합을 하고 가수분해가 진행된 다.38-42,63-78 또한 수분을 구성하는 -OH group은 쌍극자 상호작용(dipole-dipole interaction)에 의해 PA분자에 흡착되어 가수분해가 진행된다.19,27 이는 인장강도의 감소로 이어지고 시험 결과에서도 같은 경향이 관찰된다.18
또한 GF가 충전된 복합소재 내에서 수분이 흡착되면 복합소재의 인장강도와 인장탄성률 감소가 관찰되는데, 이는 수분 흡착에 의해 낮아진 매트릭스 수지와 GF 계면간의 응력 전달능력(stress transfer capability)의 감소가 GF의 보강성을 저하시키는 원인으로 알려져 있다.80
Conclusion
가수분해(hydrolysis)의 기전(mechanism) 과 측정방법, 그리고 실제 소재 제조공정에서 적용되는 분야에서의 가수분해의 이론적인 부분에 대하여 고찰하였다.
기존의 py-GC-MS의 용도는 정성적인 분석에 응용하였지만 SIM응용기법을 적용하면 정량적인 분석도 가능하다고 학계에 알려진 부분을 산업현장에서 PA66의 가수분해에 의한 degradation을 정량적인 분석을 적용하여 측정하는 방법을 최초로 적용하고 소개하였다. 이는 다음 파트에서 소개할 수분에 의한 PA소재 물성저하와의 관계에서 원인규명에 핵심적인 과학적 근거를 제시한다. 위에 예시한 가수분해 이외에도 수많은 기초적인 과학지식들이 현장에서도 응용을 통하여 더욱 많은 과학적인 발전이 확대대기를 기대한다.