Introduction
PU has excellent processability and is used in various industrial fields such as insulations, automotive interiors, adhesives, paints, and films. PU is composed of organic matter, which makes it vulnerable to fire. Most conventional way to improve the flame retardancy of PU is physical and chemical incorporation of halogen-based flame retardants which have high-cost competitiveness. However, their use has been restricted due to adverse effects on the environment and the human body.
Phosphorus containing flame retardants, which are less harmful to environment than halogen-based flame retardants, form an intumescent char during the combustion of PU. This intumescent char not only blocks the contact of combustible organic matter with oxygen, but also has the effect of preventing the transfer of activation energy needed for combustion reactions, thereby improving the flame retardancy and thermal stability of PU. The detail fire retardancy mechanisms of phosphorus intumescent additive polymer systems were reported in elsewhere.1-7
Many studies were performed to enhance the flame resistance and thermal stability of PU using phosphorus containing materials. For instance, S. W. Shin and his colleagues8 introduced melamine phosphate (MP) and Cloisite 30B in the esterification step between adipic acid and diethylene glycol to modify polyol, followed reaction with MDI to synthesize flame retardant PU. They studied the pyrolysis characteristics of the synthesized PU with TGA and reported the decomposition rate significantly reduced at high temperatures, and the residual amount also greatly increased. A. Lorenzetti et al.9 studied the effect of commercial aluminium phosphinate products on the flame retardancy of PU foams. They loaded 10 and 20 wt.% of IPA (aluminium phosphinate, 29.2 wt.% P), Exolit OP1230 (aluminium tris-diethlyphosphinate, 23.3~24.0 wt.% P), and Exolit OP1200 (mixture of aluminium tris-diethlyphosphinate and nitrogen-based synergist, 15.4~16.4 wt.% P) to PU foams. Their TGA thermogram showed that the residue weights of the PU foams increased to 20 percents by adding 20 wt.% Exolit OP1230 at 800°C in air. However, the cone calorimeter results showed that the residue weight of the PU foams decreased from 29 to 21~26% by adding the aluminium phosphinate products. O. Kwon et al.10 improved the fire-resistance of PU foam, which was made from ether-based polyol and TDI, by adding phosphorus compounds such as tetramethylene bis(ortho phosphorylurea), phosphinyl alkylphosphate ester (CR-530), resorcinol bis(diphenyl phosphate) (RDP), triethyl phosphate (TEP), etc. They studied pyrolysis characteristics of the PU foam and reported that the residual weight of the PU foam increased by 8.3 wt.% and the maximum heat release rate decreased from 378.05 kW/m2 to 359.76 kW/m2. S. Duquesne et al.11 added 40 wt.% ammonium polyphosphate (APP) to MDI base PU to raise the flame retardancy and investigated the fire retardancy of the APP-added PU in air condition. They found that the decomposition of the APP-added PU began at lower temperatures than that of virgin PU, and the carbon layer resulted from the decomposition formed the phosphocarbonaceous polyaromatic structure with the phosphorus of the APP. It was reported that the residue of the APP-added PU increased due to the phosphocarbonaceous layer that was more stable than the carbonaceous structure made from PU. Differing from many researchers using additive- and solid-type phosphorus containing flame retardants, H. S. Park and his colleagues12 studied the effect of phenylphosphonic acid (PPOA) on the flame retardancy of polyester taffeta coated with PU. They synthesized the modified polyester diol containing tetramethyl bis(ortho phosphate) and PPOA. And then they cured the polyester diol with hexamethylene diisocyanate biuret that has three NCO groups. They report that the residues of the cured polyester taffeta increased with the concentrations of PPOA. Their residual weight data were focused on the polyester taffeta and limited to temperatures lower than 400°C.
Several research focused on the effect of isocyanate structure on PU thermal stability. M. Barkini et al.13 prepared polyurethane dispersion from PTMG, DMPA, 1,4-BD, H12MDI, IPDI, MDI, and TDI. They measured the thermal stability of the casting film of the PUD using TGA in air and showed that the thermal stability increased with the order of MDI-PU > H12MDI-PU > TDI-PU > IPDI-PU. They insisted that the high thermal stability of MDI-PU and H12MDI-PU was the consequence of intense H-bonding by the symmetrical diisocyanate structure that caused more phase separation. M. V. Pandya et al.14 studied the effect of diisocyanate structure on the viscoelastic, thermal, and mechanical properties of PUs that were synthesized from ester type polyol and each of crude MDI, TDI, HDI, and IPDI. They determined the relative stabilities of the polyester PUs by comparing the PUs’ integral procedural decomposition temperatures and decomposition temperatures. They reported that the thermal stability was ordered as follows: crude MDI > TDI > IPDI > HDI.
In this work, we tried to incorporate PPOA into PU backbone structure and studied the effect of the PPOA on the thermal stability of the PUs. The incorporation of PPOA was performed by modifying each of ether type polyols (Mw=62, 106, 190, 419, 605) with PPOA. And then each of the PPOA modified polyols was reacted with 4,4′-dicyclohexylmethane diisocyanate (H12MDI) or 4,4-diphenylmethane diisocyanate (MDI). The content of PPOA in the PU was manipulated by varying the molecular weight of the polyols. The effect of the PPOA amount and isocyanate molecular structure on the thermal decomposition characteristics of the PPOA modified PU were studied in air environment.
Experimental
Phenylphosphonic acid (PPOA) (98.0%) was purchased from TCI. Ethylene glycol (EG, 99.0%,), diethylene glycol (DEG, 99.0%), polyethylene glycol 200 (PEG200, MW= 190), and polyethylene glycol 400 (PEG400, MW=419) were obtained from Daejung Chemicals & Metals Co. Ltd. Polyethylene glycol 600 (PEG600, MW=605) was purchased from Sigma-Aldrich. The molar mass of the PEG200, PEG400, PEG600 were measured as 190, 419, 604, respectively. The polyols were dried over 10 hours at 60°C before used. 4,4′-dicyclohexylmethane diisocyanate (H12MDI) (99.5%+, Covestro) and 4,4-diphenylmethane diisocyanate (MDI) (99.8%, BASF) were used without purification.
PPOA and the polyol were charged into a 4-neck round flask attached a mechanical agitator and a condenser. The PPOH/OH molar ratio was fixed to 0.5 and Tin(?)-2-ethylhexanoate was used as catalyst. Reaction temperature was maintained at 150°C. The reaction system was purged with N2. The functional groups of the prepared PPOA were semi-quantitatively analyzed using FT-IR. Table 1 lists the feed composition and hydroxy value (OHV) of each PPOA modified polyol.
PPOA-PU was synthesized from each of the PPOA modified polyols and each of two different isocyanates, H12MDI and MDI. The NCO/OH molar ratio was fixed at 1.05 for all reactions. The reaction was performed in a 4-neck round flask attached a mechanical agitator and a condenser under N2 atmosphere. The PPOA modified polyols were dried for 10 hours prior the reaction. The reaction temperature was maintained at 90°C when H12MDI was used and 60°C when MDI was used. The extent of reaction was determined by measuring NCO% and the reaction was ceased at the point that the measured NCO% was not changed. The functional groups of the prepared PPOA-PU were semi-quantitatively analyzed using FT-IR. If necessarily, small amount of DMF was added into the reactor to modulate the viscosity of the reactant-product mixture. To compare the thermal stability of PPOA-PUs with that of non-PPOA PUs, the corresponding PU, not containing PPOA, were also prepared. Table 2 and 3 list the formulations used to prepare PPOA-PUs from the PPOA modified polyol and each of H12MDI and MDI, respectively. Table 4 and 5 list the formulations used to prepare non-PPOA PUs from the polyol and H12MDI or MDI, respectively.
| Formulation of non-PPOA PU | |||||
|---|---|---|---|---|---|
| PU-1H | PU-2H | PU-3H | PU-4H | PU-5H | |
| Polyol / wt.% | EG / 18.4 | DEG / 28.0 | PEG200 / 40.8 | PEG400 / 60.4 | PEG600 / 68.7 |
| H12MDI wt.% | 81.6 | 72.0 | 59.2 | 39.6 | 31.3 |
| Formulation of non-PPOA PU | |||||
|---|---|---|---|---|---|
| PU-1M | PU-2M | PU-3M | PU-4M | PU-5M | |
| Polyol / wt.% | EG / 19.1 | DEG / 28.8 | PEG200 / 42.7 | PEG400 / 61.7 | PEG600 / 69.9 |
| MDI wt.% | 80.9 | 71.2 | 57.3 | 38.3 | 30.1 |
The hydroxyl value, defined as the weight of KOH (mg) required to neutralize the remaining acetic acid after acetylation of one gram of the reactant product mixture containing free hydroxyl groups, was determined by back titration method in excess acetic anhydride and pyridine solvent. The NCO content of the raw isocyanates and the PU products was also determined by back titrating the unreacted di-n-butylamine with hydrochloric acid after urea formation from sample’s isocyanate and excess di-n-butylamine. The detail procedures for the hydroxy value and NCO content measurements are described in KS M 0065 and KS M5969, respectively. FT-IR measurements (model: Thermo Fisher Scientific, Nicolet iS20) were performed to identify and to semi-quantitatively analyze the functional groups of PPOA modified polyols and PPOA-PU. TGA (model: Setaram instrumentaion, SETSYS Envolution, 10?/min) measurements were carried out to characterize the thermal properties of the PPOA-PUs and non-PPOA PUs under air condition. The atom contents of the phosphorcarbonaceous char, obtained after TGA analysis of the PPOA-PU, were measured using FIB-SEM/EDS (model: Thermo Fisher Scientific, Scios2, 10 kV).
Results and Discussion
Figure 1 shows the reaction scheme for modification of polyols with PPOA and followed by reaction the PPOA modified polyol with MDI or H12MDI. It is expected that PPOA modified polyol has two structures, I and II. In Step 1, P-OH group of PPOA reacted with OH group of polyol to form P-O-C group depicted in structure I and II. Figure 2 shows the IR spectra of pure EG and the PPOA modified EG mixture. Absorption peaks associated with P-OH, P-OC, and C-O stretching are shown at 1680 cm-1, 795 cm-1, and 880 cm-1, respectively. Table 6 lists the characteristic absorption peaks of PPOA, polyol, and the PPOA modified polyol.15-21
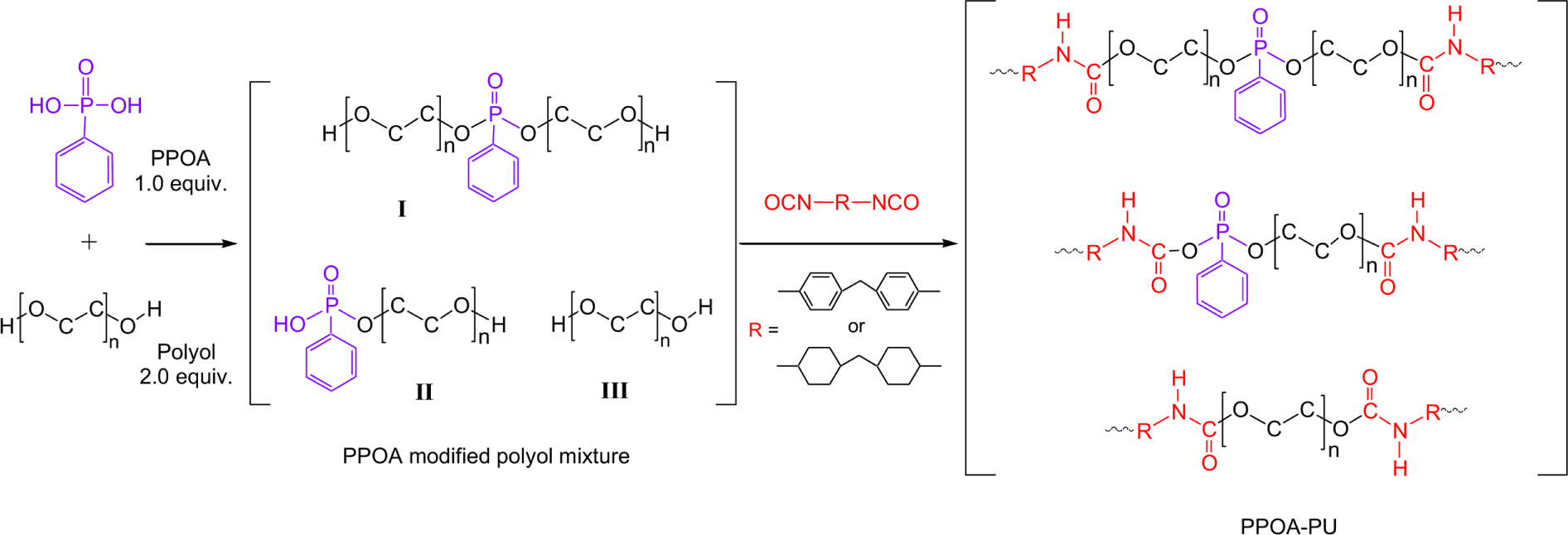
The extent of the modification reaction was determined using P-OH stretching peak (1680 cm-1) and P-OC stretching peak (795 cm-1) of the PPOA modified polyol mixture. As the reaction proceeded, the P-OH stretching absorption decreased whereas the P-OC stretching absorption increased. The reaction was ceased at the point that the P-OH stretching absorbance (1680 cm-1) of the PPOA modified polyol mixture did not vary with time. It was unable to evaluate the yield of the PPOA modified polyol, probably due to the instability of the O-P-O bond at the reaction temperature. Tang et al.22 reported the similar behavior in the modification of polyester with PPOA. They suggested that the O-P-O bond of PPOA might be significantly unstable at temperatures above 240°C. Therefore, some phosphorus of the PPOA likely dispersed in the PPOA modified polyol mixture.
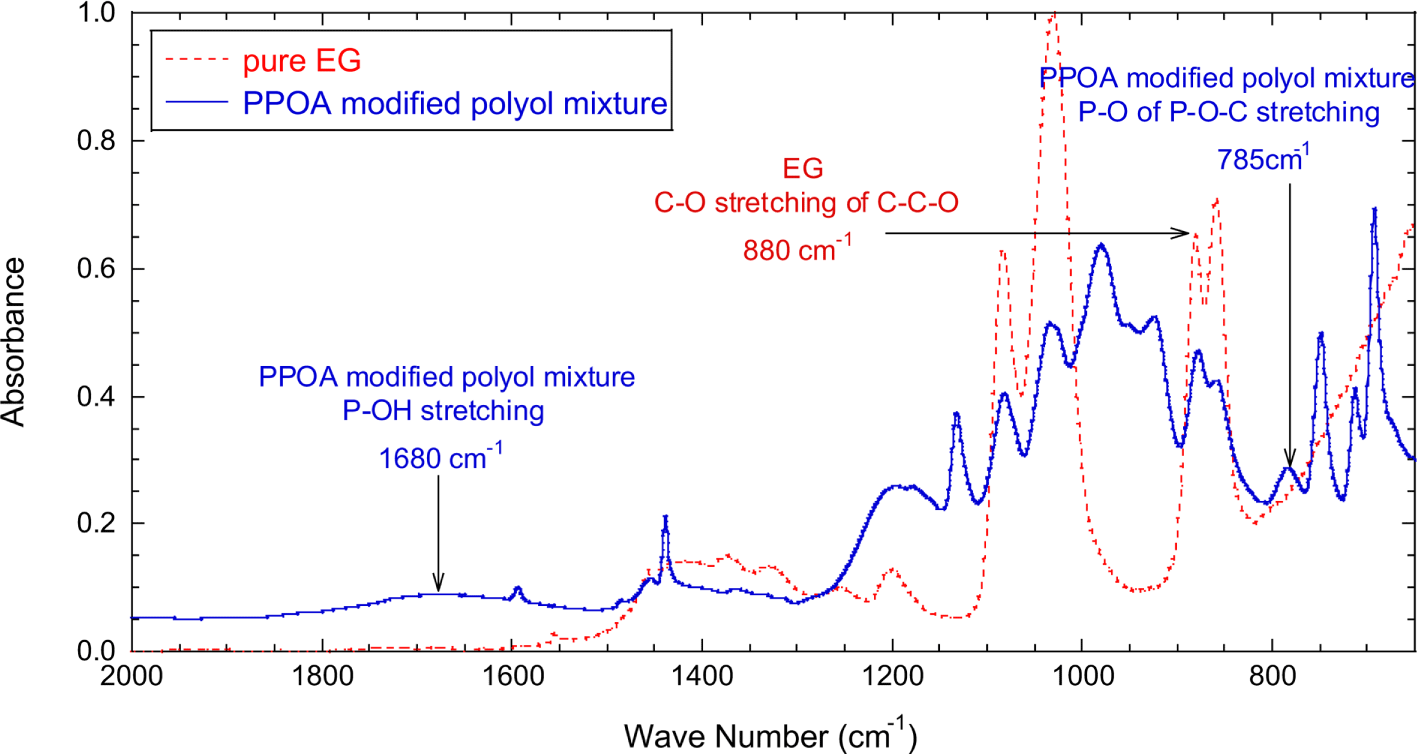
The absorbances of P-OH and P-OC stretching vibrations were normalized with the absorbance of C-O stretching (880 cm-1) of polyether polyol. The C-O group of the polyether polyol did not participate in the modification reaction and maintained its structure during the reaction. Figure 3 shows that the normalized absorbance of P-OH peak (A1680/A880) quickly decreased with reaction time till 200 minutes, then leveled off, and little changed after 500 minutes. Figure 3 also well demonstrated that the normalized absorbance of P-OC peak (A785/A880) increased quickly by 250 minutes, then leveled off, and reached equilibrium around 500 minutes. Note that the FT-IR measurements began as soon as the reaction temperature reached at 150°C. After reaction step 1, it was expected that the PPOA modified polyol (structure I and II) and unreacted polyol existed in the product mixture.
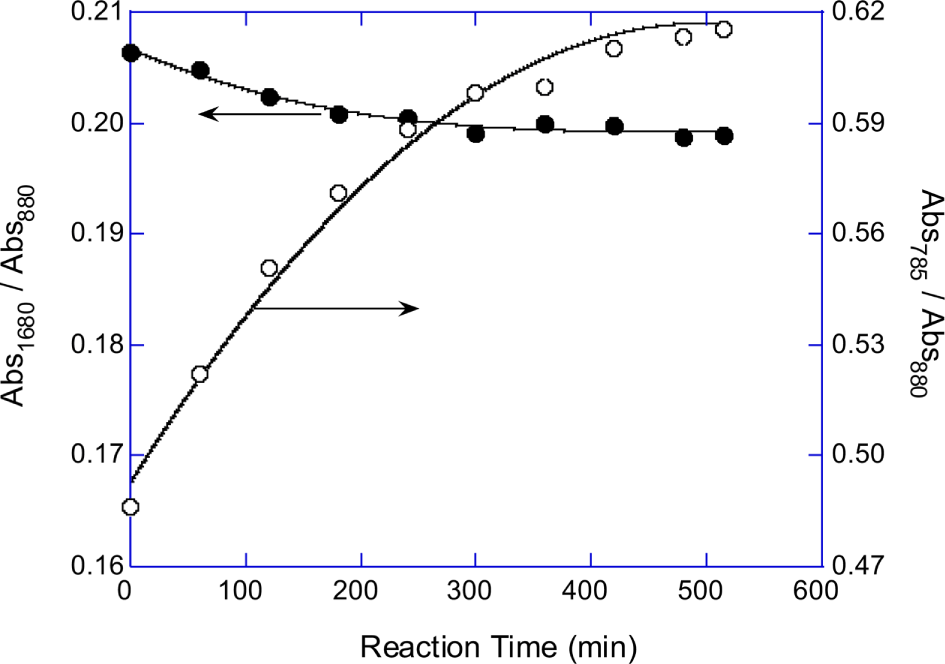
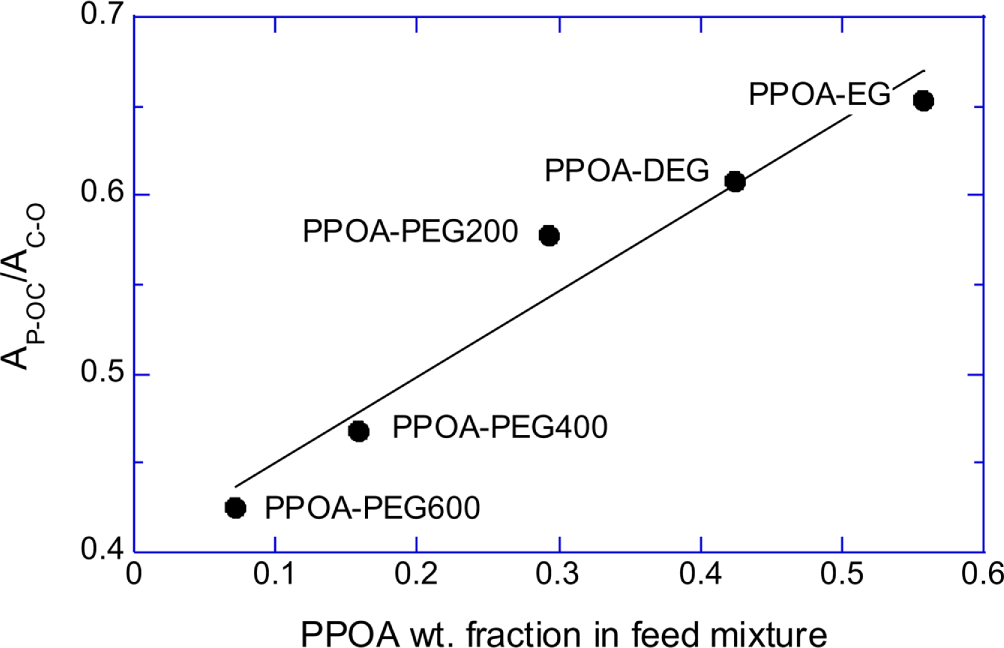
Normalized absorbances of P-OC peak (A785/A880) of the PPOA modified-EG, -DEG, -PEG200, -PEG400, and -PEG600 mixture products were plotted in Figure 4A. Figure 4B shows that the absorbance at 785 cm-1 (P-OC peak) of the synthesized PPOA modified polyols linearly related with the PPOA weight fraction in the feed mixtures. The equivalent ratio of the polyol to PPOA was fixed at 0.5 for all modification reactions. The number of C-C-O unit of polyol increases with the polyol molecular weight. Therefore the (A785/A880) values, which implicitly represent P content of the PPOA modified polyols, increased with decreasing the chain length of the polyols. Figure 3, Figure 4A, and 4B suggest that the extents of the polyol modifications by PPOA were similar regardless the chain length of the polyols.
Using the PPOA modified polyols and each of two different isocyanates, H12MDI and MDI, PPOA-PU was synthesized at the fixed NCO index of 1.05. Figure 5 shows FT-IR spectra of PU-1H-5 and PU-1M-5. The detailed formulation of PPOA-EG, PU-1H-5, and PU-1M-5 are described in Table 1 and 2. The absorption bands associated with C=O stretching (mostly H-bonded) and N-H stretching are shown at 1700 cm-1 and 3330 cm-1, respectively. Although not shown in Figure 5, as the molecular weights of the raw polyols increased from 62 (EG) to 605 (PEG600) the C=O stretching peaks of the synthesized PUs shifted to 1723 cm-1. The wavenumber shift is attributed to H-bonding of urethane C=O groups. The number of urethane C=O groups per PU chain increased as the molecular weight of the raw polyol decreased. Therefore, the more the molecular weight of the raw polyol increases, the less the shape and position of the urethane C=O peak exhibit the H-bonded C=O characteristic and shift to high wave numbers. No N=C=O stretching peak (2270 cm-1) is in the both spectra. The IR spectrum of PU-1H-5 (dashed line) shows the peak at 1452 cm-1 associated with -CH2- stretching and that of PU-1M-5 (solid line) does the peak at 1598 cm-1 associated with phenyl ring C=C stretching. The peaks at 1452 cm-1 and 1598 cm-1 were used to normalize the absorbance of the urethane C=O peak of PU-1H-5 and PU-1M-5, respectively.
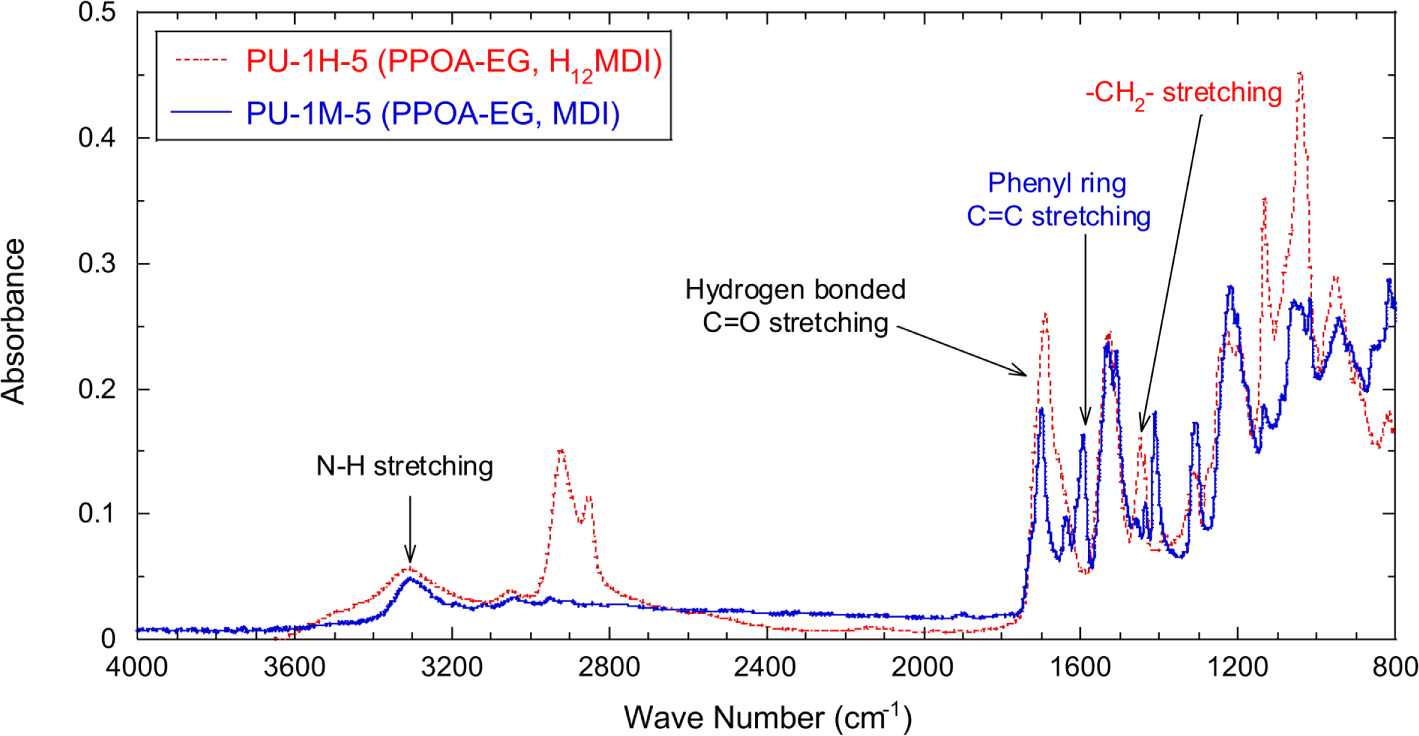
Figure 6A shows the normalized absorbances of urethane C=O peak (AC=O/ACH2) of PU-1H-5, PU-2H-4, PU-3H-3, PU-4H-2, and PU-5H-1. Figure 6B shows the normalized absorbances of the C=O peak (AC=O/AC=C) of PU-1M-5, PU-2M-4, PU-3M-3, PU-4M-2, and PU-5M-1. Figure 6A and 6B semi-quantitatively demonstrate that the number of the urethane repeat units decreased as the molecular weight of the raw polyols increased.
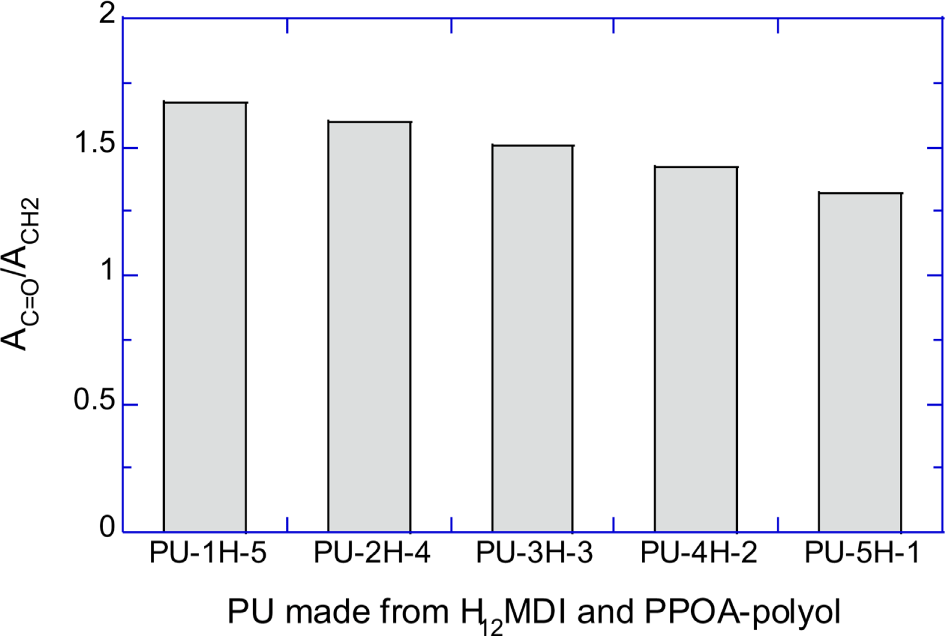
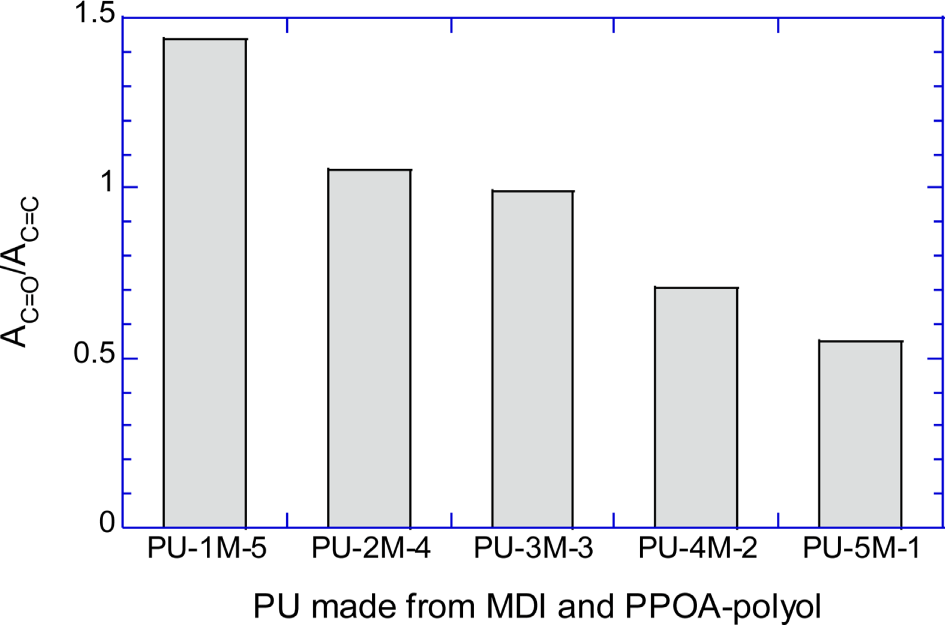
Figure 7 shows the TGA curves of the H12MDI-PUs (PU-1H-0 ~ PU-5H-0) that were prepared with H12MDI and pure polyols. Prior to presenting our experimental results, introducing the previous studies on PU thermal degradation may be helpful to understand typical thermal decomposition behavior of PU.
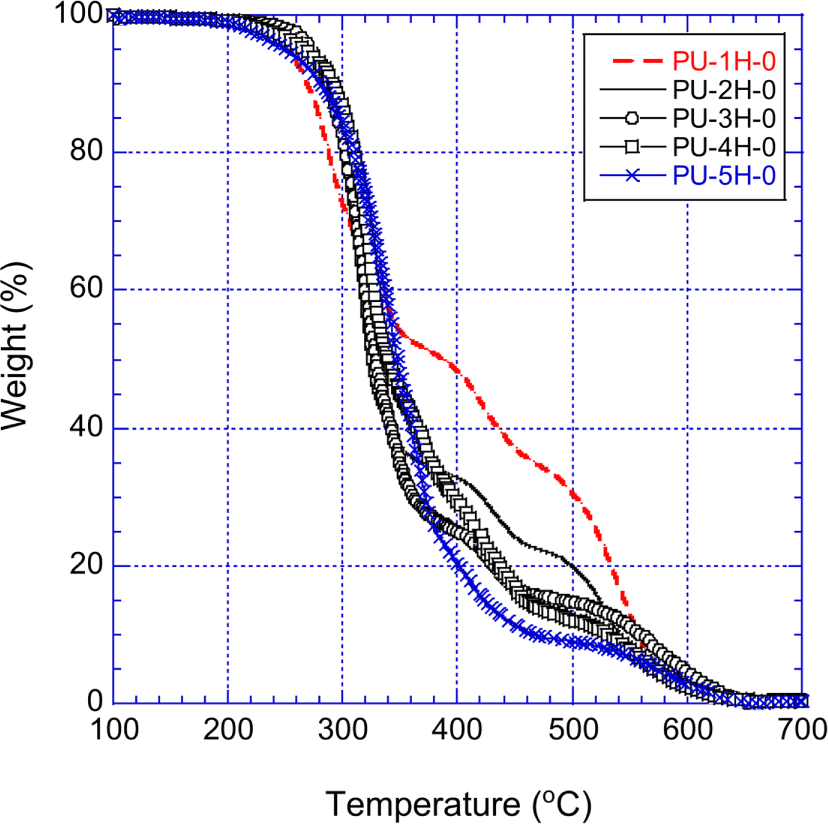
The thermal decomposition of PU began around 200°C at which mainly the urethane repeat units broke up and PU separated into the diisocyanate parts and the polyol parts. Some of the decomposed diisocyanate converted to CO2 and primary amine, then converted into urea.23,24 Urea further decomposed to produce HNCO, biuret and NH3. The biuret decomposed and converted to NH3, cyanuric acid, and ammelide with trace amount of ammeline and melamine.25-28 The generation of cyanuric acid, ammelide, ammeline, and melamine maximized at ~250°C. Ammelide and ammeline were stable at 300°C, but totally decomposed by 350°C.25,27 The first fast decomposition was observed about 250°C. With increasing temperature to 250~380°C, the polyols part predominately decomposed and produced carbonyl compounds. At this stage, the decomposed diisocyanate converted to carbodiimide and CO2. Carbodiimides were known stable up to ~500°C in air.29 The weight loss of PU moderated from 350 ~360°C due to carbodiimides that were stable up to ~500°C in air. At temperature above 550°C the rate of the second fast decomposition rerose, and the rapid weight loss reoccurred.
Figure 7 well demonstrates the typical thermal decomposition behavior of unmodified PU in air. The urethane decomposition affected differently the thermal stability of the PUs depending on temperatures. Since the urethane units of the PUs began to decompose around 200°C and the amount of urethane units increased with decreasing molecular weight of the raw polyol, between 200°C and 250°C the weight loss of the PUs was getting greater as the molecular weight of the row polyol decreased. On the other hand, the amount of ammelide, ammeline, and carbodiimide, which were originated from the decomposed urethane units, increased with decreasing molecular weight of the raw polyol. Therefore, at temperatures higher than 380°C the thermal stability (i.e., the low weight loss) enhanced with the order of PU-5H-0 < PU-4H-0 < PU-3H-0 < PU-2H-0 < PU-1H-0. At temperatures higher than 700°C, the PUs entirely decomposed in air and none of the residue exist.
Figure 8 shows the TGA curves of the MDI-PUs (PU-1M-0 ~ PU-5M-0) that were prepared with MDI and pure polyols. The residue weights of the MDI-PUs and the other PUs are listed in Table 7. The thermal decomposition behavior at temperatures below 320°C is similar to that of the unmodified H12MDI-PUs shown in Figure 7. However, between 320°C and 680°C the TGA curves show that the stability of the unmodified MDI-PUs is much higher than that of the unmodified H12MDI-PUs. For instance, at 500°C the residue weight of the unmodified MDI-PUs was 10.5~22.5 percent higher than those of the unmodified H12MDI-PUs. At 600°C, the residue weight of the unmodified MDI-PUs was 3.1~15.9 percents higher than those of the unmodified H12MDI-PUs. Except PU-1H-0 and PU-1M-0, the residue weight difference of the two corresponding unmodified PUs (e.g., the residue wt.% of PU-3M-0 minus the residue wt.% of PU-3H-0) increased with the amount of the raw MDI. Since incorporation of aromatic rings in the PU backbone significantly improved thermal stability, the more PU had the phenyl groups in its structure, more the PU was thermally stable. At temperatures higher than 680°C, the unmodified MDI-PUs were entirely decomposed and remained no residue as unmodified H12MDI-PUs.
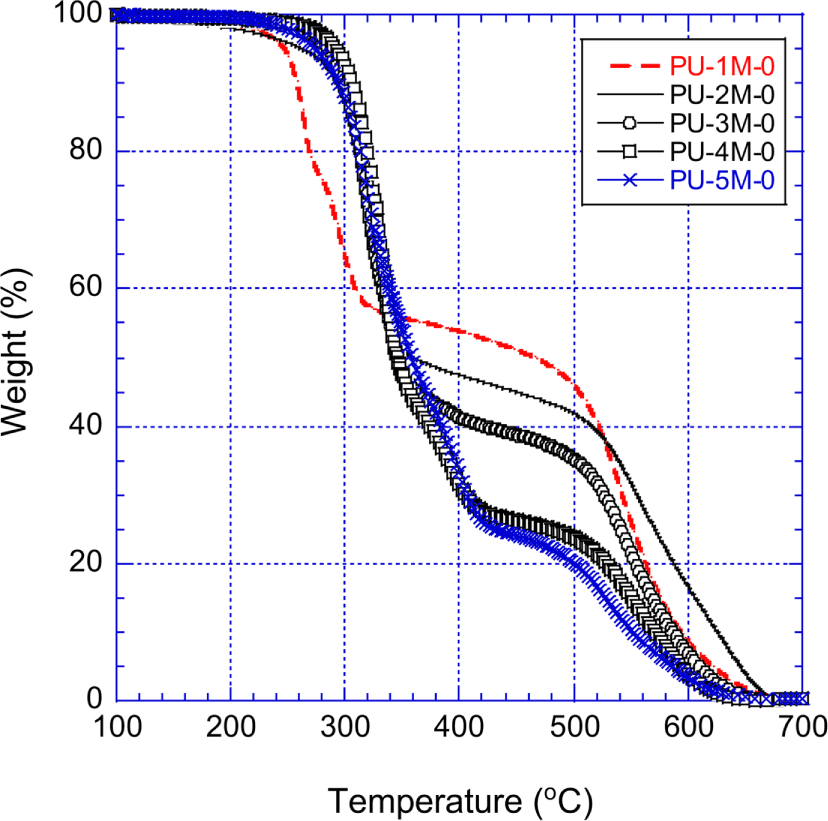
Figure 9 shows the TGA curves of the PPOA-H12MDI-PUs that were prepared with H12MDI and PPOA modified polyols. The thermal decomposition behavior of the PPOA-H12MDI-PUs is fundamentally similar to that of the H12MDI-PUs at temperatures below 350°C. However, above 500°C the PPOA-H12MDI-PUs were more thermally stable than the unmodified H12MDI-PUs. At 500°C, the residue weights of the PPOA-H12MDI-PUs were 5.2~8.4 percents higher than those of the H12MDI-PUs. At 700°C, the PPOA-H12MDI-PUs remained 1.5~17.5 wt.% residues while the unmodified H12MDI-PUs did no residues. This enhanced thermal stability of the PPOA-H12MDI-PUs is attributed to the intumescent char barrier effect originated from the PPOA moiety of the PPOA-PUs. As referred above,11,23-29 the thermal decomposed intermediates including phosphoric acid were generated between 200°C and 350°C and then made the phosphor-carbonaceous layer with the decomposed oxygen-carbon matters of the PPOA-PUs. The phosphor-carbonaceous barrier inhibited heat and O2 contacting the decomposing materials (i.e., ammelide and ammeline, carbodiimide etc.). Note that the second fast decomposition of the PPOA-H12MDI-PUs were hardly seen. At temperatures higher than 400°C, the residue weights of the PPOA-PUs followed the order of PU-1H-5 > PU-2H-4 > PU-3H-3 > PU-4H-2 > PU-5H-1. This order coincided the PPOA amount of the PPOA-H12MDI-PUs. The PPOA-PUs’ thermal stability increased with the amount of PPOA which played a significant role in forming phosphorcarbonaceous layer.
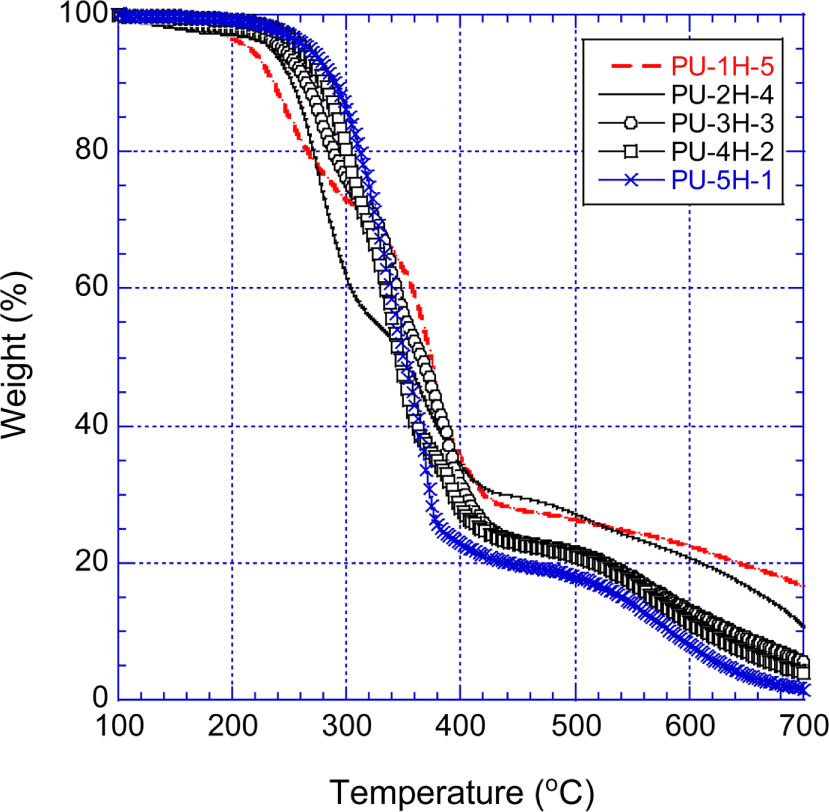
Figure 10 shows the TGA curves of the PPOA-MDI-PUs that were prepared with MDI and the PPOA modified polyols. Between 200°C and 300°C, the residues of the PPOA-MDI-PUs were less than those of the unmodified MDI-PUs (shown in Figure 8). The higher weight loss of the PPOA-MDI-PUs can be explained as follows: PPOA decomposition occurred at relatively low temperatures. The concentration of the PPOA increased with decreasing the molecular weight of the raw polyol of the PUs. Figures 9 and 10 demonstrate that the more the molecular weight of the raw polyol are low, the more the weight loss of the PUs increases.
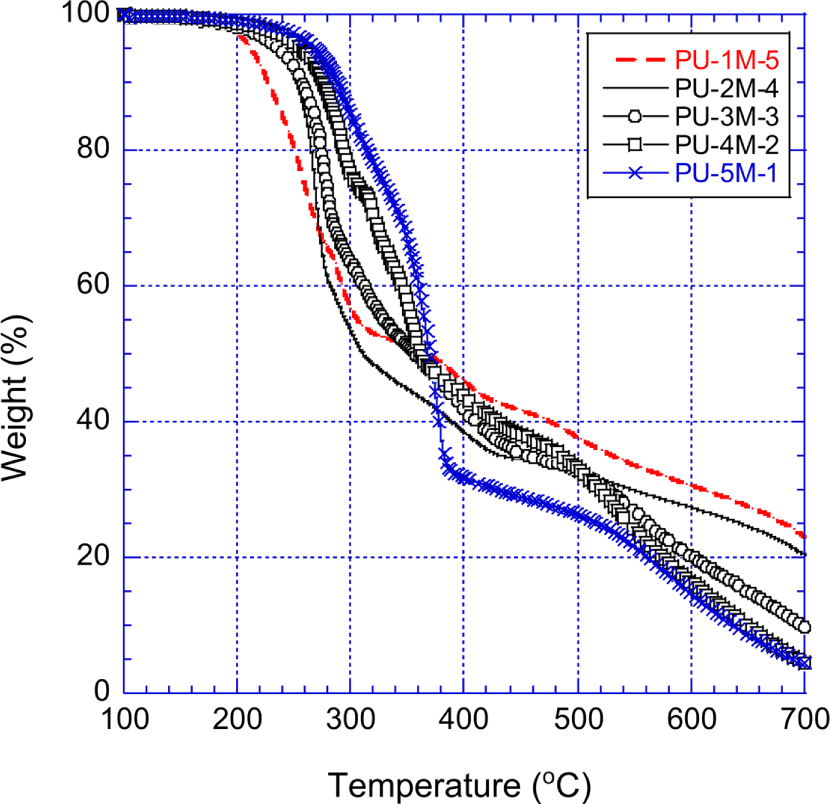
Up to 450°C the unmodified MDI-PUs, especially PU-1M-0 and PU-2M-0 manifested higher thermal stability than the PPOA-MDI-PUs. However, above 550°C the PPOA-MDI-PUs exhibited the most excellent thermal stability among the PUs used in this study. This high temperature stability is attributed to PPOA and phenyl group of the PPOA-MDI-PUs. Between 350°C and 500°C, the thermal stability of the PPOA-MDI-PUs was improved mainly due to the phenyl ring structure of MDI. The phenyl group of MDI is liable to form polyaromatic hydrocarbon that act as precursors of carbonaceous char. At temperatures higher than 550°C, the phosphorcarbonaceous char, made from the PPOA and the decomposed carbon matters, played significant role to enhance the thermal stability of the PPOA-MDI-PUs. At 700°C, the PPOA-MDI-PUs remained 4.4~23.0 wt.% residue whereas the PPOA-H12MDI-PUs and the unmodified MDI-PUs did 1.5~17.5 wt.% residue and no residue, respectively. The second fast decomposition was not observed during the PPOA-MDI-PUs decomposition.
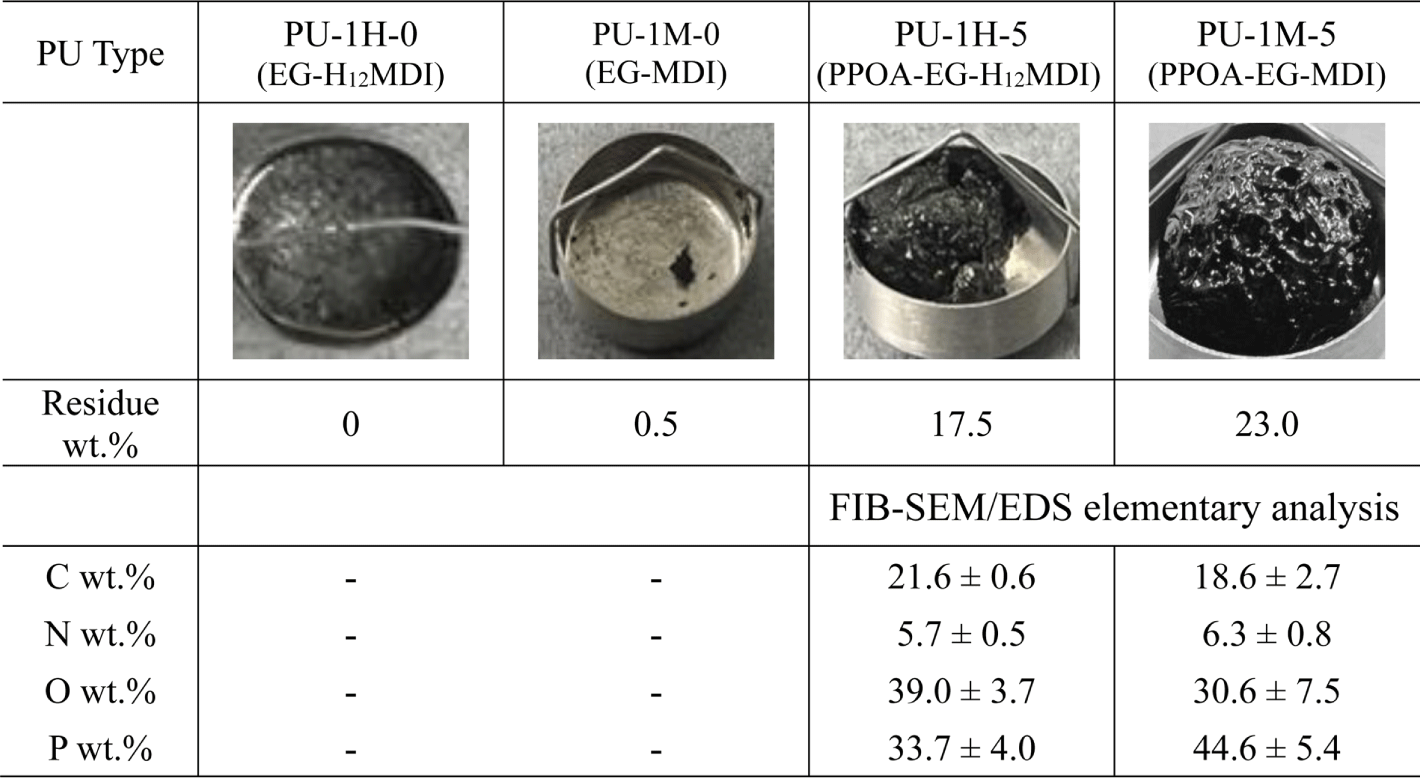
Figure 11 shows the char images of the PU residues after TGA measurement up to 700°C in air. PU-1M-5 can efficiently form the intumescent phosphorcarbonaceous char since MDI’s phenyl groups easily convert to the polyaromatic hydrocarbon precursors. PU-1M-5 char has bulkier shape and higher residue wt.% (23.0 wt.% vs 17.5 wt.%) than PU-1H-5 char. The FIB-SEM/EDS analysis of the PU chars shows that the residue of PU-1M-5 contained 18.6 wt.% C and 44.6 wt.% P while that of PU-1H-5 did 21.6 wt.% C and 33.7 wt.% P. The FIB-SEM/EDS results suggest that the intumescent phosphorcarbonaceous char of PU-1M-5 inhibits the outgassing of phosphorus matters that is more volatile than carbon matters. The uniform and uncollapsed char of PU-1M-5 entrapped the decomposed phosphorus matters within the phosphorcarbonaceous char barrier. As a result, the P content of PU-1M-5 char was higher than that of PU-1H-5.
Conclusions
The molecular structure of MDI and H12MDI are similar to each other but MDI has aromaticity. Between 350°C and 500°C, the thermal stability of MDI-PU was higher than those of H12MDI-PU due to MDI’s phenyl structure. The PPOA moiety of the PPOA-PU formed phosphorcarbonaceous char layer with the carbon matters generated during the thermal decomposition of the PU. The effect of the PPOA on the thermal decomposition was different depending on temperatures. At temperatures lower than 500°C, the thermal stability of the unmodified MDI-PUs was higher than that of PPOA-H12MDI-PUs since PPOA breaking occurred at relatively low temperatures and some of the phosphorus matter released form the PU. At temperatures above 700°C the PPOA-H12MDI-PU and the PPOA-MDI-PU remained more amounts of residues, representing higher thermal stability, whereas unmodified PU did none. Especially, the PPOA-MDI-PUs remained 4.4~23.0 wt.% residue at 700°C and were most stable at temperatures above 550°C. This high temperature stability of the PPOA-MDI-PU resulted from the synergic effect of MDI’s phenyl groups and the PPOA of the PU on forming the phosphorcarbonaceous char. PPOA-MDI-PU formed uniform and uncollapsed intumescent phosphorcarbonaceous char that inhibited the decomposing materials contacting with heat and O2. Incorporating 8.9~26.0 wt.% PPOA (1.8~5.1 wt.% P) into PU greatly enhances PU thermal stability especially at temperatures higher than 550°C. The molecular weight of polyols used for most industrial applications is higher than 3000. To find the possible applications, our study on the thermal stability of the PPOA modified PU will be extended to the PEGs with high molecular weights and OH functional groups greater than 2.0.