Introduction
Polyurethanes (PUs) are used in a variety of applications, such as upholstery foams and automotive seats, refrigerators and building insulations, textiles, coatings, and adhesives.1-3 Due to its widespread use, a lot of PU waste is generated, but there is no suitable recycling method in economical point of view. PU waste has typically treated by landfill or incineration.
Recycling of PU can be classified into mechanical and chemical methods. The mechanical method recycles PU scrap waste in a bonded or a compressed form after pulverizing it into powder. The pulverized PU powder is used as a filler. There are five mechanical recycling methods: rebinding,4,5 regrinding,6,7 adhesive pressing,8 compression molding,9 and injection molding.10 Among the five methods, regrinding and rebinding have attracted attention in the PU industry due to the low processing cost.4,5,7
The regrinding method consisted of two main process steps.7-12 For instance, flexible polyurethane foam (F-PUF) scrap waste is first grinded into fine powder using roll milling. The powder is then mixed with new polyols and reformed by reaction with isocyanates. In the milling step, the powder size should be small enough not to affect the cell structure and the properties.7 The suitable powder size was reported as 50-200 μm depending on the applications.11 Diessel et al.12 described the regrinding method of thermosetting PU soft foam to fabricate various high-strength sheet structures for airborne sound insulation sheet in noisy spaces. PU form was pulverized to granules and compressed at 50~150 bar and 180~210°C without addition of polymeric binder.
The first step of rebinding recycle method is similar to that of regrinding method. PU foam scrap waste is broken down into fine particles of an appropriate size depending on the application. The shredded powder is treated with a binder to convert to a rebound foam block. Then the foam blocks are molded to have final product shape.11 Kliwer and Kittel13 had reported the fabrication method of composite foams from PU foam flakes of different sizes and/or PU foam flakes of different material compositions using PU dispersion binder. They used an aqueous binding mixture of NCO prepolymers containing polyamides with primary or secondary bound amino groups. Mixture of PU foam flakes and the dispersion binding agent were molded typically at 4 bar and 140~160°C. The molded PU rebinding foam can be used for sound insulation in vehicle, machinery construction, and building construction.
The chemical recycling method of PUs uses thermal energy to break the urethane repeat units that link the polyol and isocyanate segments.14-18 The previous studies on the chemical recycling method are summarized as follows. In general, thermal decomposition of PU begins at around 200°C. Conventional recycling chemical methods heat PU under non-oxygen conditions to 200~300°C with decomposing agent such as water, light alcohol, and NH3. The decomposing agents also serve as a solvent to dissolve the decomposing polyol and isocyanate (or amine). Depending on the type of decomposing agent, the chemical recycling methods are classified into hydrolysis, glycolysis, and ammonolysis.
The glycolysis method uses a diol with a high boiling point such as ethylene glycol or diethylene glycol. To accelerate the decomposition, a basic catalyst, usually KOH or NaOH, is used in the glycolysis method. However, del Amo et al. reported that by-products were produced by prolonged exposure to high temperatures. The polyol produced by the glycolysis was obtained in the transesterified carbamate form rather than the original polyol form used to synthesize the rigid PU.19
Mishra et al.20 studied the hydrolysis of PU foam waste of unknown molecular structure at 15~800 psi and 100~220°C using an autoclave apparatus. They reported that the PU decomposition was independent of pressure between 13.7 and 55.2 bar, but the extent of decomposition of PU foam was strongly influenced by temperature. At 130, 150, 200, and 220°C, the hydrolysis conversion reached at 40, 50, 90, and 100%, respectively. Motokucho et al.21 studied the thermal decomposition of PU in the mixture of water and CO2. They measured the extent of PU decomposition with variations of the decomposition time, temperature, and the mixture pressure. They found that the temperature of the mixture affected decomposition of PU foam, but the pressure did not. After decomposition for 12 hours under the mixture pressure of 80 bar, the PU foam decomposed to less than 10% at 120~160°C, but 43% at 180°C and 63 % at 190°C.
To the best of our knowledge, no studies have been conducted to determined how fast the decomposition of PU occurs in real time and how the type of isocyanate used to prepare the PU affects the decomposition temperature. In this study, we tried to find the effect of isocyanate type on the decomposition temperature of PU hydrolysis in subcritical water medium. PU samples were prepared with two aromatic diisocyanates, one unbranched aliphatic diisocyanate, and two cyclic aliphatic diisocyanates. Hydrolysis of the PU samples was performed in water at pressures up to 250 bar. In addition, the dissociation time of PU hydrolysis was measured in real time by directly observing the hydrolysis reaction to be performed in a pressure view cell. Information on the decomposition temperature and required time of PU hydrolysis in subcritical water medium is expected to be useful for designing the hydrolysis process of PU recycling plants.
Experimental
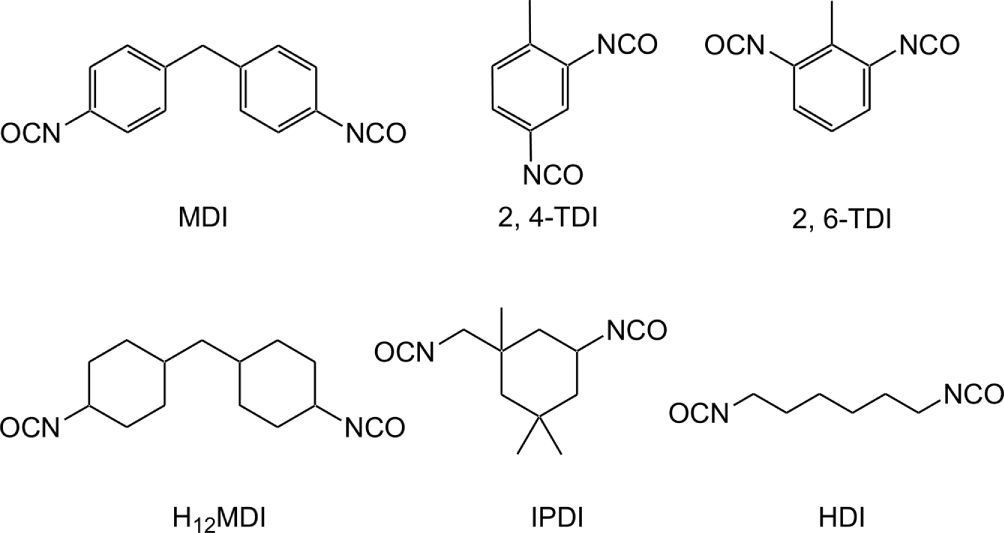
PU samples were synthesized from 1,4-Butanediol (BD, 99%, Sigma-Aldrich) and three different types of diisocyanate: two aromatic diisocyanates (4,4'-methylene diphenyl diisocyanate (MDI) and methyl phenylene diisocyanate (TDI)), one unbranched aliphatic diisocyanate (hexamethylenelene diisocyanate (HDI)), and two cyclic aliphatic diisocyanates (4,4'-methylene dicyclohexyl diisocyanate (H12MDI) and isophorone diisocyanate (IPDI)). MDI (purity, 98%) and TDI (mixture of 2,4-TDI 80% and 2,6 TDI 20%) were purchased from Acros Organics and Alfa Aesar, respectively. H12MDI (> 90%), IPDI (> 99%), HDI (> 98%) were obtained from TCI. Dimethyl sulfoxide (DMSO, 99.9%, Acros Organics) was used as reacting solvent. All raw materials were used as received. The molecular structures of diisocyantes used in the experiment are shown in Figure 1.
PU samples were synthesized by solution polymerization with DMSO as a solvent. The molar ratio of diisocyanate to BD was kept 1.05 for all PU samples. The amount of DMSO was modulated depending on the type of diisocyanate. After diisocyanate and BD mixture was agitated for 1 hour at room temperature, 0.2 wt.% of dibutyltin dilaurate (DBTDL), based on reactant weight, was added the reactant mixture. The mixture was reacted for 48 hours at 80°C, and then dried for 24 hours at 70°C in vacuum oven. Table 1 lists the detail composition of each PU sample.
The prepared PU samples were hydrolyzed in a high-pressure reactor described in detail elsewhere.22 Typically 1.7 g of PU sample and 6.6 g of water were loaded into the cell. The pressure of PU-water mixture was measured with Heise gauge (Dresser Ind., Model CM, 0-2068.4 bar, accurate to within ± 2.1 bar). The temperature of PU-water mixture was measured using platinum-resistance thermometer (hand A therm Technology Co., Class A) and a digital thermometer (Omega, DP9800-RTD, accuracy ± 0.05°C). The mixture temperature was maintained within ± 0.5°C. The mixture inside the reactor was viewed on a video monitor using a camera coupled to a borescope placed against to the outside of the view window. The mixture of the decomposed material and water was a single phase or two plashes, depending on the isocyanate type used to prepare the PU samples. After the hydrolysis of MDI and H12MDI based PU samples, BD- and water-rich upper phase and amine-rich solid bottom phase were obtained. Aqueous single phase, containing BD and amine, was obtained from TDI, IPDI, and HDI based PU hydrolysis.
The fourier-transform infrared spectroscopy (FT-IR) measurements of the PU samples and the hydrolyzed materials were performed at room temperature with a Thermo Nicolet iS20 FTIR-ATR spectrometer. Single beam spectra of the samples were obtained after averaging 64 scans between 4000 and 650 cm-1 with a resolution of 4 cm-1.
Proton nuclear magnetic resonance (1H-NMR) analysis was performed to identify the structure of the hydrolyzed products. The hydrolyzed materials of PU sample were dried for 24 hours at 70°C in vacuum oven and dissolved to be approximately 1 w/v% in DMSO-d6. The 1H-NMR spectra of amines, obtained by the hydrolyzed, were determined using Agilent MR400 DD2.
Results and Discussion
Solid PU samples (S1~S5), filled with water in the high-pressure reactor, were hydrolyzed at 70~250 bar and 199~237°C. It was found that pressure did not affect the decomposition behavior between 70~250 bar, so the hydrolysis was performed at a fixed pressure of 140 bar. This independency of MDI-PU decomposition on pressure was also reported by Motokucho et al.21 When the temperature of the mixture of PU and water reached the specific decomposition temperature, the hydrolysis of the PUs was completed in 1 to 5 minutes and the solid PUs were completely dissolved in water. Table 2 lists the decomposition temperatures of the PUs with the melting or glass transition temperatures.
| Sample No. | Tg (°C) | Tm (°C) | Decomposition* Temperature (°C) |
|---|---|---|---|
| S1 (MDI-PU) | 68.4 | 165.8 | 237.0 |
| S2 (TDI-PU) | 42.0 | - | 199.0 |
| S3 (H12MDI-PU) | 95.1 | 178.6 | 218.0 |
| S4 (IPDI-PU) | 41.2 | - | 218.0 |
| S5 (HDI-PU) | - | 177.3 | 220.0 |
The decomposition temperature of the PU samples increases in the following order: TDI-PU (199°C) < H12MDI ≈ IPDI ≈ HDI (218~220°C) < MDI-PU (237°C). This temperature order is related to the electron donating ability of the group to connected to the nitrogen of the urethane unit. The raw isocyanate structures before urethane reaction are shown in Figure 1. Nolan et al.23 reported that the aromatic ring (MDI) donates less electrons to the nitrogen of the urethane unit of PU than the aliphatic ring (H12MDI and IPDI) and aliphatic HDI. Because the electron density of the nitrogen of the urethane unit of MDI-PU is relatively low, MDI-PU has low reaction activity. Therefore, the MDI-PU (S1) decomposed at the highest temperature among the PU samples. In the case of TDI-PU, we supposed that the para-positioned urethane of TDI-PU was first decomposed into primary amine and BD, and then the primary amine attached to the phenyl ring donated electrons to the nitrogen of the ortho-position urethane to improve the second decomposition. TDI-PU was decomposed at the lowest temperature of 199°C.
At temperatures above the melting or glass transition temperature before decomposition, the PU samples became viscous and stayed as two-phase mixture with water. At the decomposition temperature, the viscous PU in the two-phase mixture was hydrolyzed and converted into a yellowish transparent homogeneous phase, indicating that the molecular weight of the decomposed product from the PU sample was sufficiently low to be soluble in water. As the temperature decreased, some of the hydrolysis product from S1 (MDI-PU) and S3 (H12MDI-PU) fell out of the solution and solidified, while TDA, IPDA, and HDA maintained a homogenous solution due to their high solubility in water. Figure 2 shows that how the shape and phase of the PU samples changed in sub-critical water during the hydrolysis.
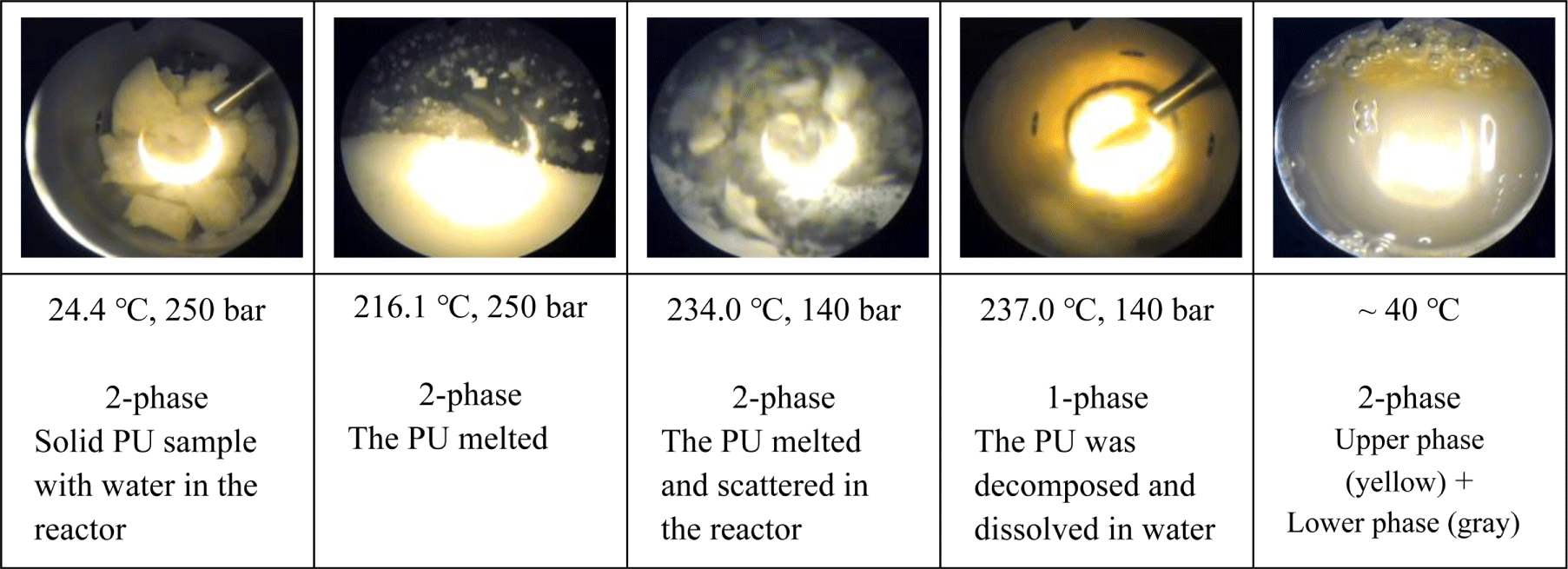
At room temperature, the hydrolysis products of MDI- and H12MDI-PU samples (S1, S3) consist of a solid lower phase and a liquid upper phase rich in BD and water. The hydrolysis product of TDI-PU, IPDI-PU, and HDI-PU sample (S2, S4, S5) formed an aqueous single phase. Figure 3 shows the hydrolysis products of S1 (MDI-PU) and S2 (TDI-PU), which exist in two phases and a homogenous phase, respectively.

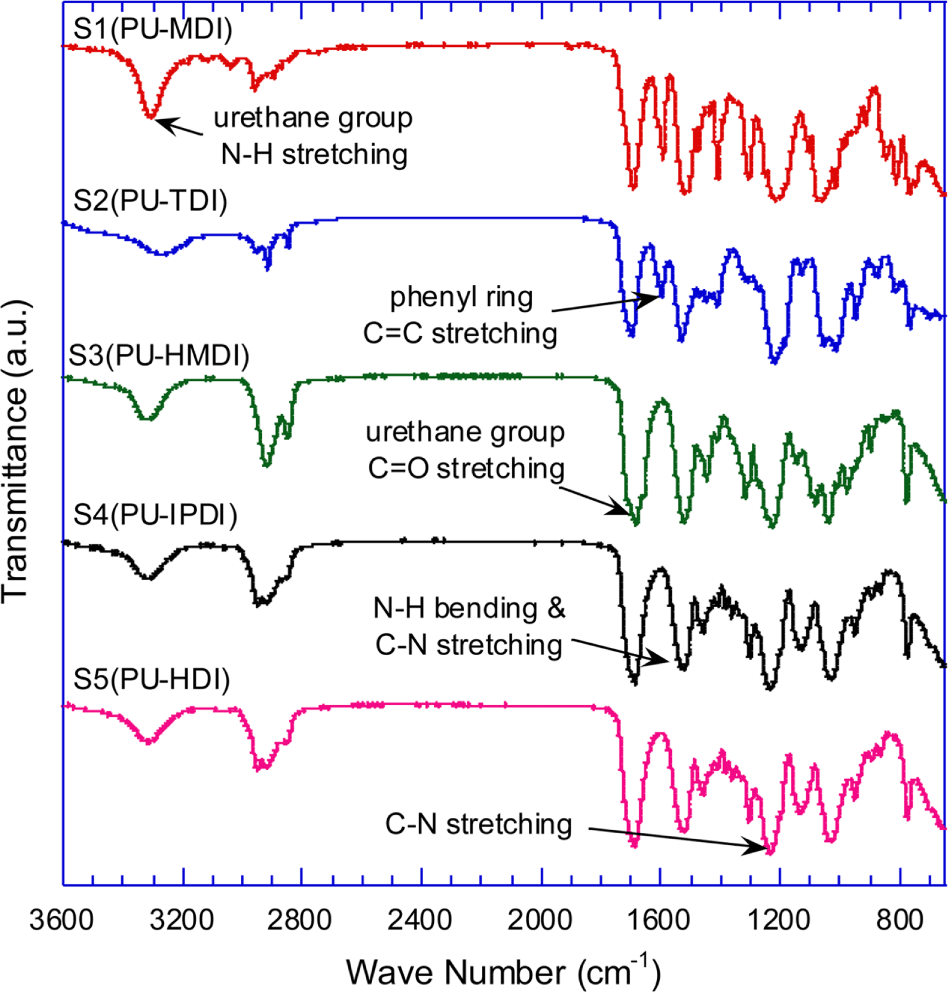
Figure 4 shows the IR spectra of the synthesized PU samples, S1~S5. In all the spectra, N=C=O stretching peak (2270 cm-1) was not shown while urethane carbonyl stretching peak (1690 cm-1) was. Phenyl ring C=C stretching peak appears at 1598 cm-1 in the spectra of PU sample (S1 and S2) made from aromatic isocyanate, MDI and TDI.
The IR spectra of S1 (MDI-PU) and its hydrolysis products are shown in Figure 5. The hydrolysis products were recovered as a liquid and a solid that formed respectively upper and low phase in the high-pressure reactor. To identify the molecular structure of the hydrolysis product, IR spectra of pure BD and methylene diphenyl diamine (MDA) are also presented for reference in Figure 5. The IR spectrum of BD shows the peaks at 3400~3200 cm-1. (H-bonded O-H stretching), ~2900 cm-1 (C-H stretching), and 1030~1050 cm-1 (primary alcohol C-O stretching). The typical characteristic peaks at 3440 cm-1 (primary amine N-H asymmetric stretching), 3410 cm-1 (primary amine N-H symmetric stretching), 1625 cm-1 (NH2 deformation), and 1500 cm-1 (phenyl ring C=C stretching) are shown in MDA IR spectrum.
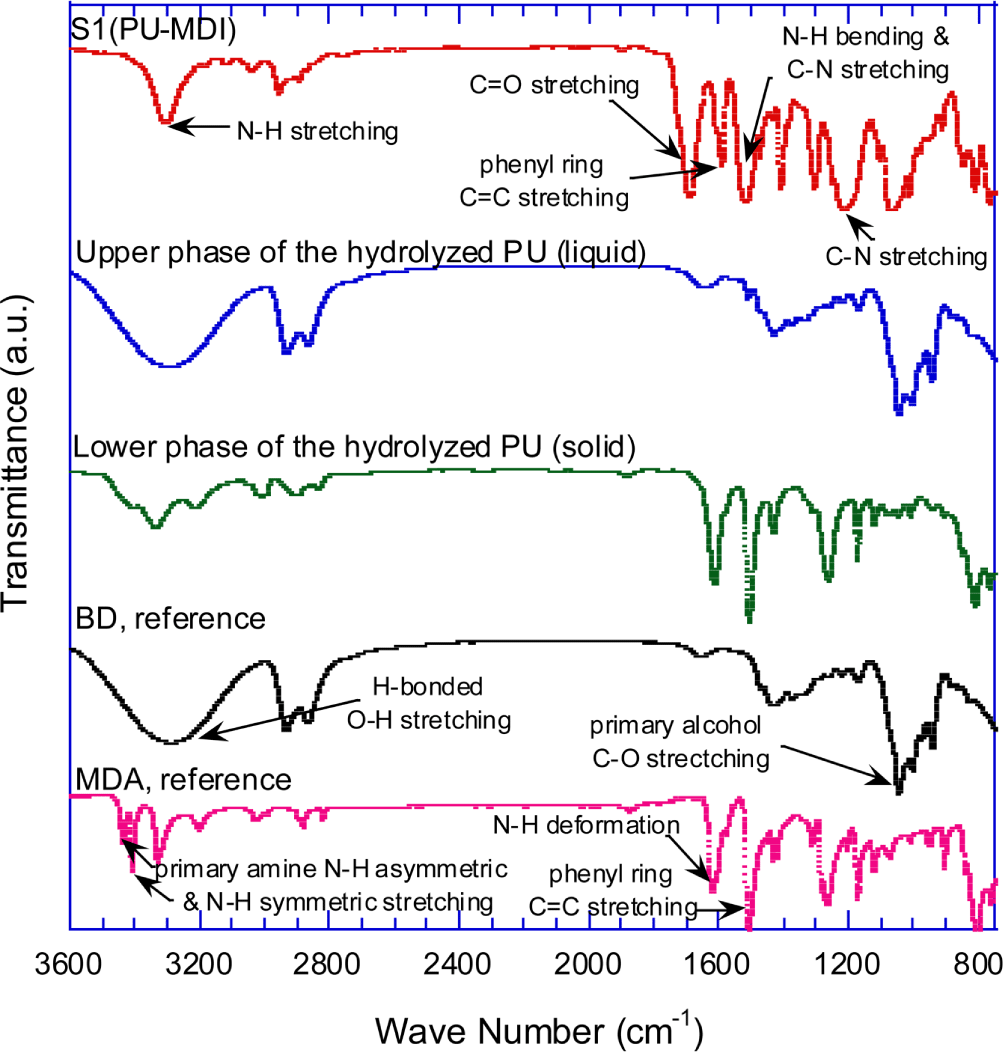
Figure 5 shows that the IR spectrum of the upper-phase (liquid) is exactly same as the spectrum of BD. The IR spectrum of the lower-phase (solid) is almost identical to those of MDA between 1650 and 800 cm-1, while the spectrum between 3450 and 3100 cm-1 shows the characteristic peaks of BD and MDA. Compared to the intensity of BD, the peak intensities of the lower-phase spectrum are weak at 3450~3100 cm-1, suggesting that a little amount of water or BD remains in the recovered solid even after drying for 24 hours at 70°C in vacuum oven. From FT-IR spectral data, it is inferred that the lower-phase solid is a diamine with essentially the same molecular structure as the diisocyante used to synthesize the PU sample, except that the N=C=O group is replaced with NH2.
Figure 6 shows the 1H-NMR spectra of the upper liquid phase and the lower solid phase of the hydrolysis products of S1 (MDI-PU). For reference, the spectra of pure MDA and BD are presented with their molecular structures. The 1H-NMR spectrum of MDA shows peaks at 6.78~6.80, 6.45~6.47, and 4.78~4.80 ppm that are assigned to the resonance of a-, b-, and c-hydrogen of MDA, respectively. The hydrogen peak of primary amine (d) is shown at 3.54~3.55 ppm. In the 1H-NMR spectrum of BD, the resonance peaks of r-, s-, t-hydrogen are shown at 4.36~4.39, 3.35~3.39, and 1.40~1.45 ppm, respectively.
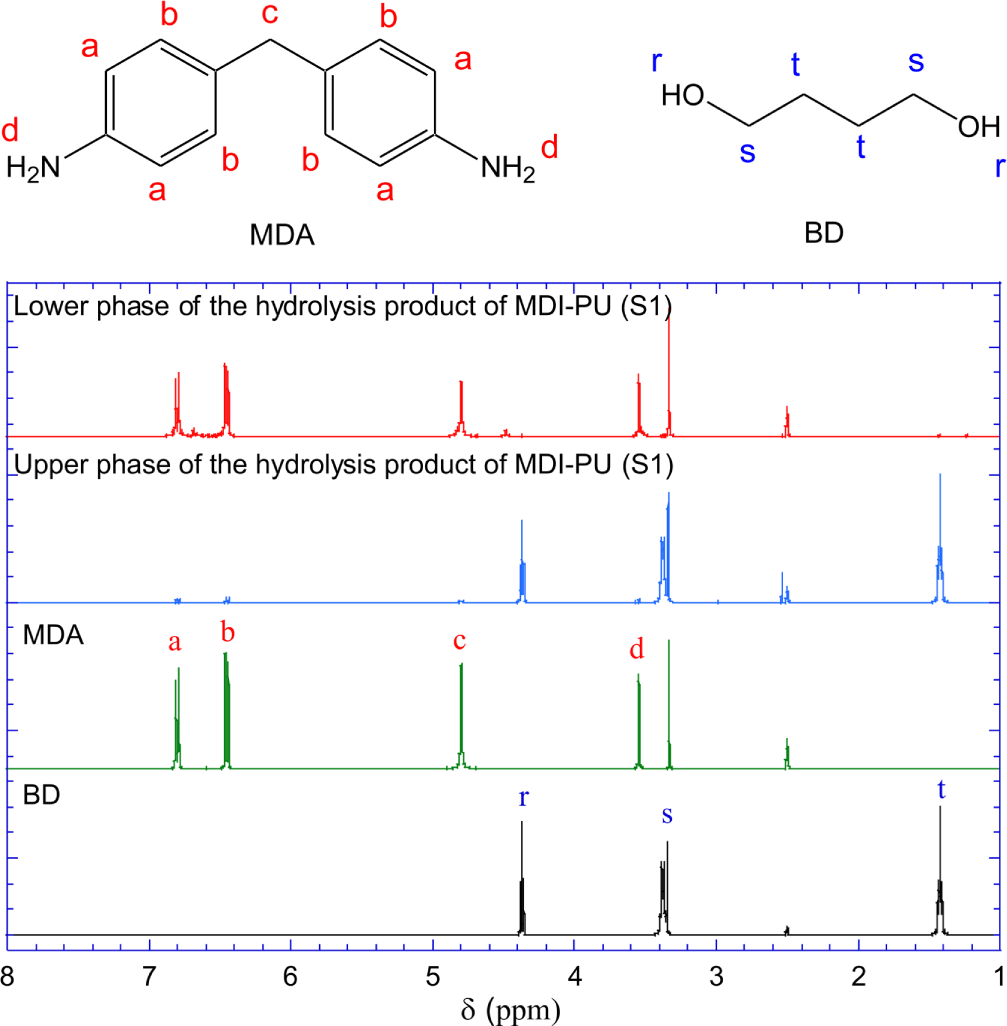
The 1H-NMR spectrum of the solid phase of the hydrolysis products is identical to the spectrum of pure MDA. Both spectra show a peak at 3.3~3.35 ppm, assigned to H2O hydrogen. No hydrogen peaks of BD are shown in the spectrum of the solid phase of the hydrolysis products. The 1H-NMR spectrum of the upper liquid phase is also identical to the to the spectrum of pure BD.
The FR-IR and 1H-NMR spectra indicate that MDI-PU (S1) hydrolyzed to BD and MDA which formed the upper liquid phase and lower solid phase, respectively, at room temperature.
Conclusions
We investigated the hydrolysis of PUs that were synthesized from different diisocyanate: MDI, TDI, H12MDI, IPDI, and HDI. Through the direct observation of PU decomposition behavior in subcritical water, it was found that the PUs were hydrolyzed within 5 minutes at their specific decomposition temperature. The decomposition temperature increase in the order of TDI-PU (199°C) < H12MDI ≈ IPDI ≈ HDI (218~220°C) < MDI-PU (237°C). This temperature order is assumed to be due to the electron donating ability of the group to linked to the nitrogen of urethane. The aromatic ring (MDI) is less electron donating than the aliphatic rings (H12MDI and IPDI) and aliphatic HDI. For TDI-PU, the para-position urethane of TDI-PU was preferentially decomposed into primary amine and BD. Afterwards, the primary amine attached to the phenyl ring donated electron to the nitrogen of the ortho-position urethane, thereby improving the decomposition.
We expect that our finding on the hydrolysis behavior of PU, including the decomposition time and the effect of isocyanate type on decomposition temperature, could contribute to the development of more economical recycling methods for PU waste.