



Introduction
Silica has been one of the most important reinforcing filler materials not only for synthetic rubber, but also for natural rubber due to a number of advantages, such as high tear strength and tensile strength, reduced heat buildup and abrasion resistance to rubber composites.1,2 Although conventional silica offers many advantages, the abundant silanol group on the surface of silica could result in strong silica-silica attraction by hydrogen bonding.3-5 Hence, the conventional silica-reinforced rubber usually exhibited high aggregation and uneven dispersion in the rubber matrix due to strong filler-filler interaction. This interaction can lead to a low efficiency of silica particles in improving the mechanical properties of the rubber products. Additionally, the drawbacks of applying silica in rubber usually exhibits high energy consumption because of the serious aggregation of silica particles, environmental pollution during the operation process, and respiratory disease as the abundant silica is dispersed into the air during production.6,7 Hence, many efforts have been made to solve these problems and to improve the reinforcement efficiency of silica in the rubber industry.
In recent years, the sol-gel process of tetraethoxysilane (TEOS) has been developed to obtain silica particles inside the rubber matrix instead of directly mixing the solid silica with the rubber matrix. The advantage of this method is that it could open another way for improving the dispersibility of silica in the rubber matrix.8 Based on this method, different polymer systems, such as styrene-butadiene rubber (SBR),9 butadiene rubber (BR),10 acrylonitrile-butadiene rubber (NBR),11 natural rubber (NR)12 and epoxidized NR13 have been reported previously. Although the base rubber latex displayed catalytic properties for the hydrolysis of TEOS, the conversion and the reaction time usually lasted for a long time.1,14,15 Hence, here the acid catalyst was first used for the hydrolysis of TEOS and then blended with the SBR latex. The gel composite of silica-reinforced in SBR matrix could then be formed quickly at a certain temperature, and this could save a lot of time during the preparation process. In addition, due to the in situ sol-gel process, the silica dispersed into the air could apparently be decreased, and this could also reduce the risk of respiratory disease during the production.
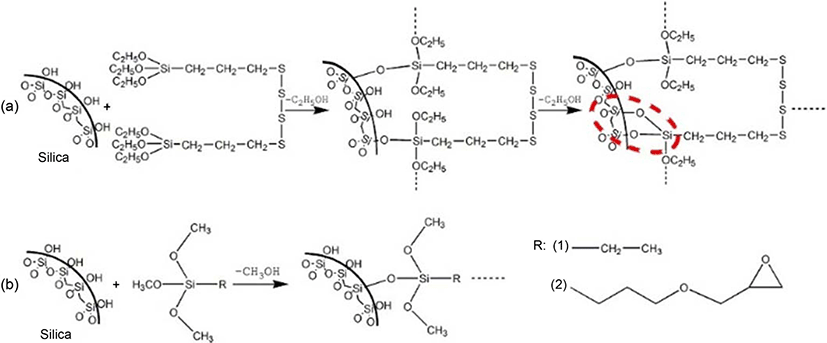
Except for the above in situ sol-gel preparation method of silica in rubber, silane coupling agents, such as bis-(3-triethoxysilyl propyl) tetrasulfane,16 alkyltrialkoxysilanes17 and iso-butyltriethoxysilane18 are usually applied to modify the surface of the silica in order to improve silica dispersion and enhance the mechanical properties of the rubber. Hence, in this paper, the silica sol was firstly synthesized by hydrolyzing of TEOS over a nitric acid catalyst. Then the silica sol was mixed with a certain amount of SBR latex at room temperature. Subsequently, the different contents of silica-reinforced SBR solid composites was obtained after oven-drying. The preparation process is summarized in Figure 1. Additionally, in order to improve the dispersion of silica in the SBR matrix, silane coupling agents such as bis[3-(triethoxysily)propyl]tetrasulfide (TESPT), (3-glycidyloxypropyl)trimethoxysilane (GPTS), and ethyltrimethoxysilane (ETS) were applied in the above system. Finally, the structure of solid silica-reinforced SBR is characterized and the mechanical properties of silica-reinforced SBR with or without silane coupling agents are systematically discussed.
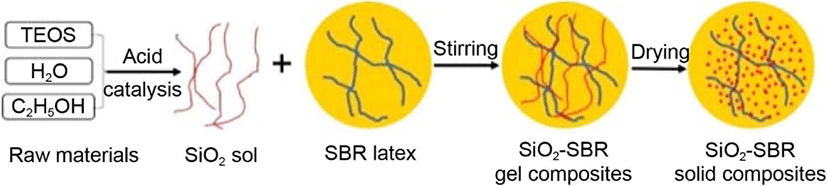
Experimental
Commercial styrene-butadiene rubber latex 1502 was purchased from GemHoo Company, Korea. Tetraethyl orthosilicate (TEOS), absolute ethanol and sulfur were obtained from Daejung Chemicals & Metals Co., Ltd.. Nitric acid was purchased from Duksan Pure Chemicals Co., Ltd.. Acetic acid and zinc oxide were purchased from Samchun Pure Chemicals Co., Ltd.. N-cyclohexyl-2-benzothiazolyl sulfonamide (CBS) and 2,2’-dibenzothiazolyl disulfide (DD) were purchased from Tokyo Chemicals Industry Co., Ltd.. Bis[3-(triethoxysily)propyl]tetrasulfide (TESPT), (3-glycidyloxypropyl)trimethoxysilane (GPTS), and ethyltrimethoxysilane (ETS) were purchased from Sigma-Aldrich Corporate.
Silica sol precursor was synthesized following the literatures.19,20 Briefly, ethanol and deionized water were added to the solution of TEOS. Then a certain amount of nitric acid was added and the pH value was kept at about 4.5. The molar ratio of the TEOS, ethanol, deionized water and nitric acid was 1:3.8:6.4:0.085. The silica sol was obtained by stirring the solution at 70°C for 3 h in a water bath.
The silane coupling agents, such as bis [3-(triethoxysily) propyl] tetrasulfide, (3-glycidyloxypropyl) trimethoxysilane, and ethyltrimethoxysilane, modified samples were prepared by the same molar ratio of TEOS, ethanol, deionized water and nitric acid as that of the samples without the silane coupling agents. After the silica sol was formed, the amount of silane coupling agents was fixed at 8 wt% (with regards to the amount of TEOS) added into the above system. Subsequently, the silane coupling agents modified samples were obtained after being stirred at 70°C for another 3 h in the water bath.
The synthesis of SBR and silica composites was obtained via the following process: The different amount of as-prepared silica sol was added to 40 mL of ethanol and 40 mL of deionized water. The solution was then poured into 175 mL of SBR latex after ultrasonic for 10 min. Finally, the above mixture was further stirred at room temperature until the gel was formed. The obtained mixture gel was dried in an oven at 60°C for about 24 h. The different amount of TEOS is summarized in Table 1.
The above dried samples were blended with zinc oxide, CBS, DD, stearic acid and sulfur on a two-roll mill at about 40°C. The contents of the composition are shown in Table 1. Finally, the vulcanized samples were fixed in a 1 mm mold under the pressure of 15 MPa at 160°C (the curing time (t90) for all the samples from the RPA test are shown in Table 2) on a heating press machine (Auto Hydraulic Press Type, Ocean Science). Subsequently, the mechanical properties of as-prepared samples were measured in the following procedure.
The morphology of the samples after the tensile test was carried out on a FE-SEM with EDS (JSM-7500F, JEOL Ltd. Japan). The valence of Si was performed on an X-ray photoelectron spectroscopy (K-alpha, Thermo. U.K.). FT-IR spectra of the samples were measured on a Perkin Elmer Spectrum 100S. Thermal properties of composites were conducted on a thermorgravimetric analyzer (TGA, Perkin Elmer 4000) by heating from 30 to 800°C at a rate of 20°C/min under the nitrogen flowing of 20 cm3/min. The cure/vulcanization characteristic of as-prepared samples were measured by a rubber process analysis (RPA) (RPA-V1, U-CAN DYNATEX INC.). The minimum torque (ML), maximum torque (MH), scorch time (ts2), and optimum cure time (t90) were determined by the above RPA. The cure rate index (CRI) was used to evaluate the cure rate of the rubber, and it was calculated by the following equation:
The strain sweep was also performed on the above RPA. The shear storage modulus was recorded for each strain at 60°C according to the ASTM D 6204-97. The hardness of as-prepared samples was obtained by a shore durometer type A according to the ASTM D 22-40. The tensile strength test was measured three times on a Tinius Olsen H5KT-0401 testing machine at a speed of 500 mm/min according to ASTM D412. The samples were made of a dumb-bell shape with the dimensions of 25 mm × 6 mm × 1 mm after the vulcanization on the heating press machine.
Results and Discussion
Figure 2 shows the SEM graphs of the fracture surface for the neat SBR, the different contents of silica in the SBR matrix and the silane coupling agents modified silica in the SBR matrix after the tensile test, respectively. As can be seen from the graphs, the fracture surface of the samples seemed smooth when the contents of TEOS were from 20 to 40 phr. During the increase of the TEOS contents to 60 phr, the fracture surface became rough and uneven (sample (E) and (F)). For the case of sample (G), (H) and (I), these samples were modified by TESPT, GPTS, and ETS silane coupling agents based on the same amount of TEOS as sample (E), respectively. Though the fracture surface for sample (G), (H) and (I) was not as even as sample (B), (C) and (D), their roughness clearly improved compared with that of sample (E), which showed the same TEOS contents and was without the modification of silane coupling agents. In addition, the Si element mapping graph for sample (B) to sample (I) were exhibited in the bottom right corner of each graph, respectively. The bright color seemed to be enhanced with the increasing of the TEOS contents, meaning that the aggregation of silica was obviously enhanced, especially for sample (E) and sample (F). Subsequently, the same content of TEOS as sample (E) was chosen for the addition of the silane coupling agents and checked for the dispersion of silica in the SBR matrix. The graphs of TESPT, GPTS, and ETS modified silica in SBR matrix are shown in (G), (H) and (I). It seemed that the bright color became lower and the silica dispersion was improved compared with sample (E).

In order to make sure the TEOS was converted to silica during the reaction process, the narrow scan of the Si 2p XPS measurement was carried out and shown in Figure 3. As can be seen in the figure, a neat SBR of sample (A) was also performed as compared with sample (E), which showed 50 phr of TEOS. The peak shown at 101.59 eV for sample (E) could ascribe to the Si 2p binding energy, and indicated the formation of silica during the reaction process. This binding energy was also consistent with the findings in previous literatures.21-23
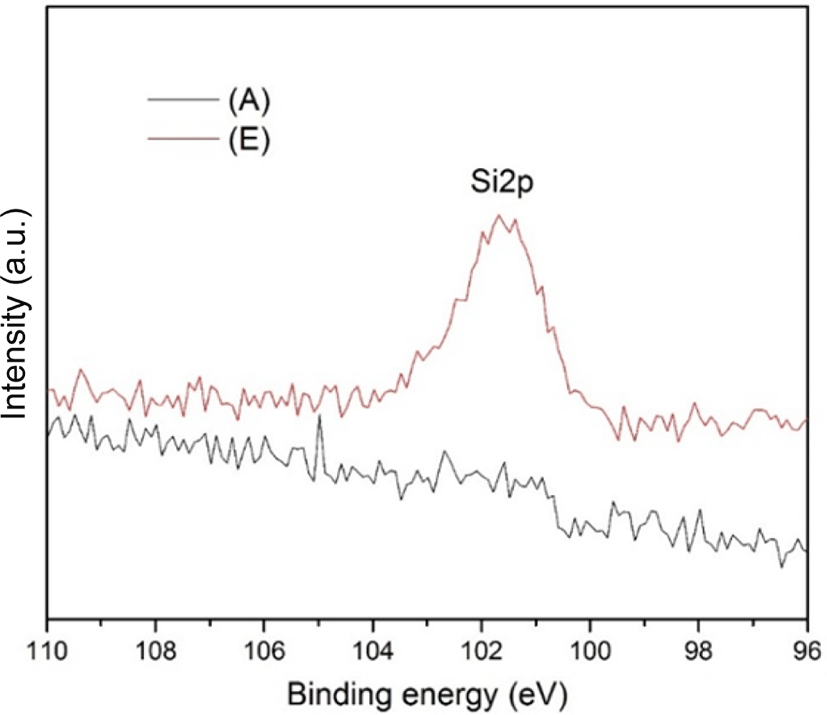
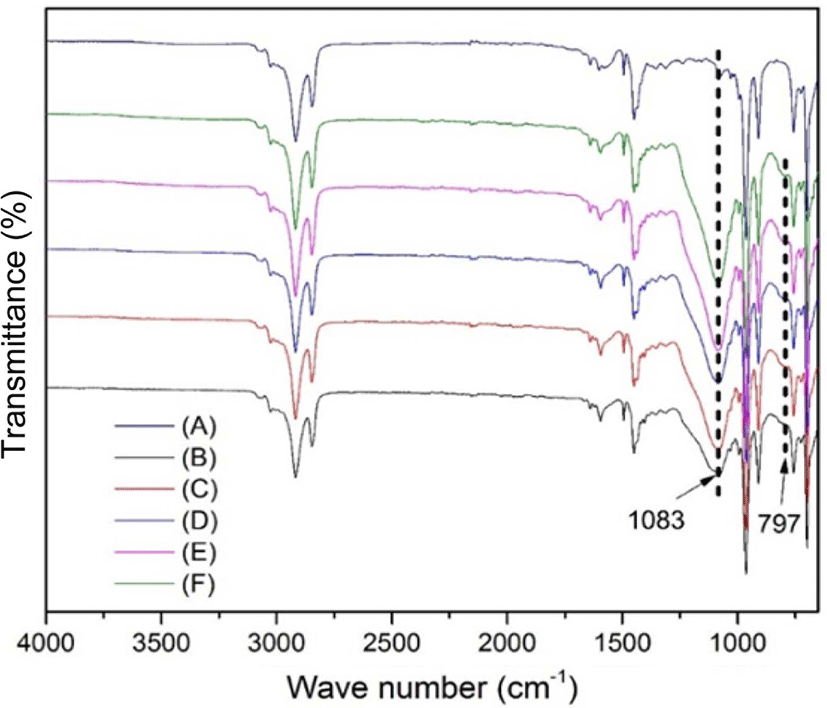
Figure 4 shows the FT-IR spectra of neat SBR and different contents of silica-reinforced SBR. As can be seen, the peaks at 2915 and 2845cm−1 are attributed to the methylene asymmetrical stretching vibration and the symmetrical stretching vibration, respectively.24,25 The absorption peaks at 1451 and 1495cm−1 belong to the skeletal vibration of the benzene ring of styrene. The absorption peaks at 700 and 967cm−1 correspond to the C-H deformation vibrations of the cis-1,4 units and trans-1,4 units of butadiene. The absorption peaks at 911cm−1 are attributed to the C-H deformation vibration of the vinyl structure of butadiene, and the absorption peak at 1639cm−1 belongs to the C=C stretching vibration of butadiene. Besides, the peak at 1083cm−1 arises from Si-O-Si asymmetrical stretching vibration, and the absorption peak at 797cm−1 is the symmetrical stretching vibration of Si-O.26
The thermal stability for the different contents of silica in the SBR matrix is determined by the TGA in Figure 5. In order to eliminate the combination effect of thermal and thermo-oxidative degradation in presence of oxygen, the thermogravimetry analysis was performed in nitrogen atmosphere. As observed in Figure 4, the residue for sample (D) to sample (F) was increased due to the increasing of silica contents. The temperature region of mass loss from about 400°C to 650°C was caused by the decomposition of SBR. The curves in the left bottom exhibited the differential TG curves and indicated the decomposition temperature of each sample. For sample (B), the decomposition temperature was about 490°C. With the increasing of the silica contents, the decomposition temperature moved to a higher temperature, about 501°C for sample (C). Subsequently, the decomposition temperature showed a slight decrease from 498°C to 491°C for sample (D) to sample (F). This may be due to the aggregation of silica in the SBR matrix. In other words, the interaction between the silica and SBR in sample (D), (E) and (F) may be not as strong as that in sample (C).
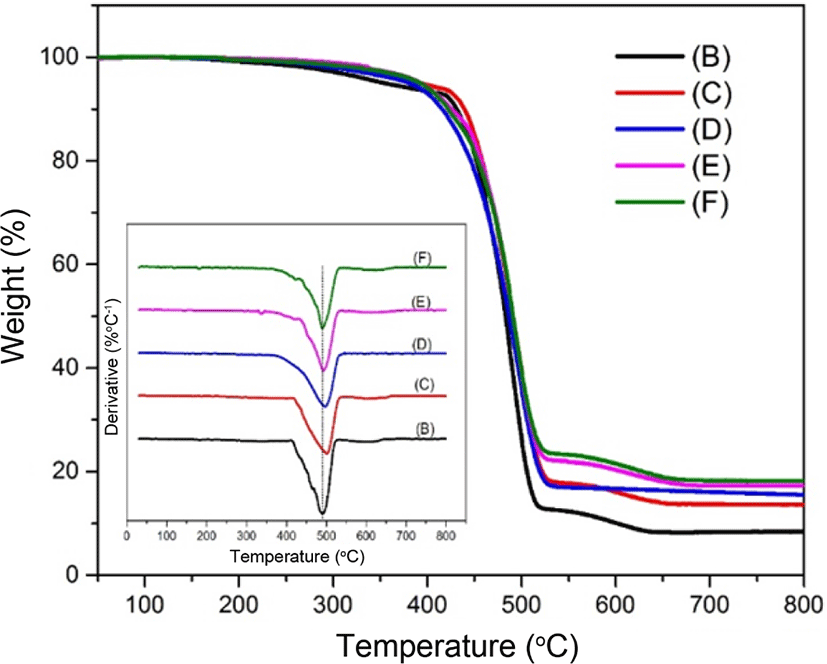
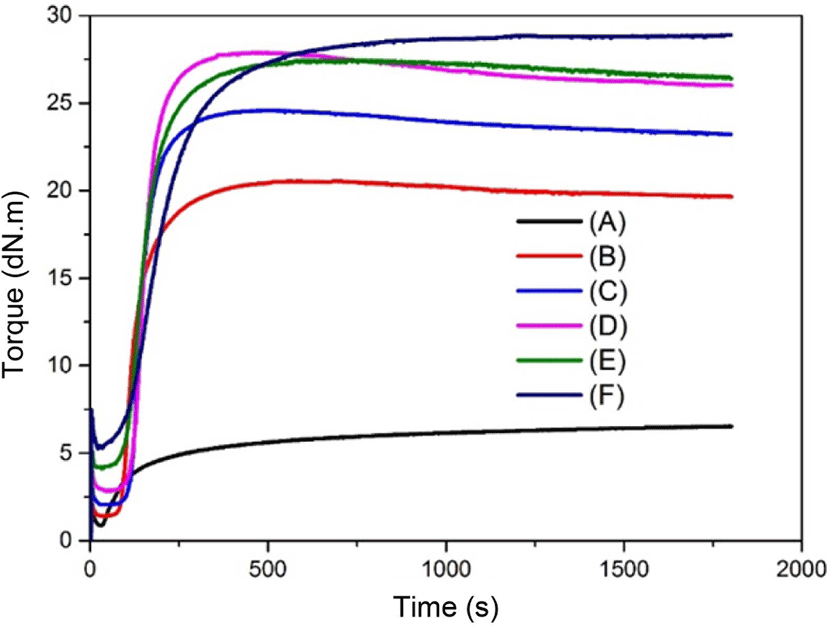
Table 2 shows the curing characteristic of neat SBR and different contents of silica in the rubber matrix. As can be seen, the maximum torque (MH) and minimum torque (ML) of the neat SBR were very low, and the CRI was just 8.64 min−1. For the non-modified silica, both the maximum torque and minimum torque of the prepared samples increased with the increasing of TEOS contents from 20 to 60 phr. This was attributed to the reduced mobility of silica in the rubber matrix.27,28 In other words, the crosslink density of the high contents silica was better than the crosslink density of low contents silica in the rubber matrix. The minimum torque value indicates the viscosity of the system. Sample (F) showed the highest value of the minimum torque (ML=5.24 dNm) mainly due to the aggregation and the high viscosity of the neat silica. This meant the movement for the rubber molecules was more difficult with the increasing of the effective network chain density. The cure time (t90) for sample (B) to (F) increased with the increasing of the silica contents gradually. However, the cure rate index showed an opposite trend, indicated the more contents of silica in the rubber matrix was not beneficial for the cure rate index. In addition, it was found that ts2 for as-prepared samples did not have a significant effect when there was no silane coupling agents added to the system. This result also showed that CRI for as-prepared samples was reduced with the increasing of silica contents. This may be owing to the adsorption of Zn complex and curatives on silica surfaces.29
The surface chemistry of the reinforcing filler influenced the cure parameters of the rubber. Cure traces of as-prepared samples are shown in Figure 6. As can be seen in the figure, the value of minimum torque during vulcanization increased with the increasing of silica contents. This is mainly due to the increased effective silica contents enhanced by the interaction among silica particles.30,31 Furthermore, the value of maximum torque for the above samples also showed the same trend, indicated the more contents of silica could also improve the interaction of silica in the rubber matrix.
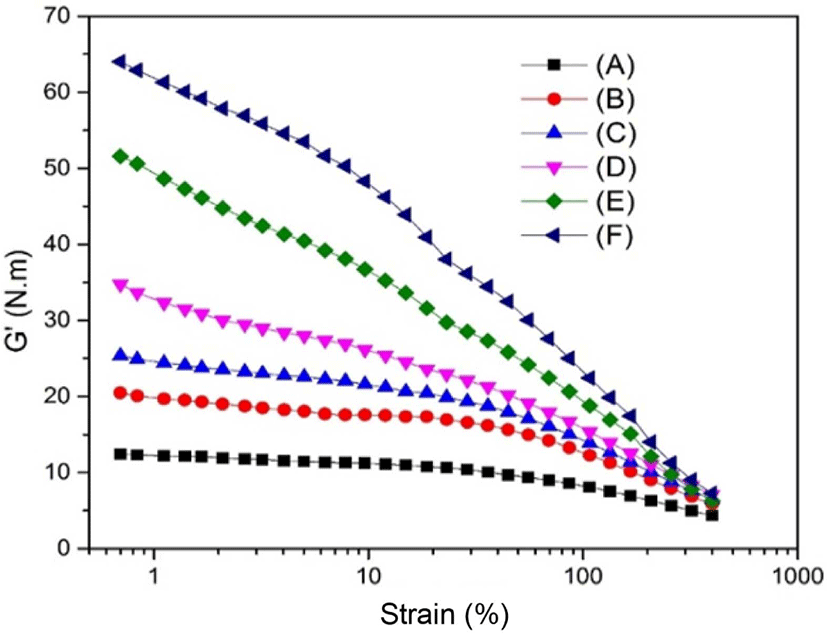
The dynamic rheological property of the SBR with different contents of silica was carried out on an RPA. The strain amplitude dependence of the storage modulus (G’) of as-prepared samples is usually to explain the filler network. There are usually two kinds of filler networks in a filler-filled rubber. One is formed by filler-filler direct contact (filler-filler contact), and another is formed by polymer chain anchored between neighbor fillers (filler-rubber network).32,33 Usually, the filler-filler network is harmful to the dynamic mechanical properties of the sample, while the filler-rubber network is beneficial to the dynamic mechanical properties of the sample. RPA measurements could exhibit the silica network in the system, and the interfacial interaction between silica and SBR matrix. As can be seen in Figure 7, the G’ decreased with the increasing of the strain amplitude for all the above samples. This result could be explained by the breakdown of aggregated secondary network among filler particles or the aggregation formed by van de Waals-London attraction forces, and this has already been illustrated by Payne. Generally, the Payne effect could be illustrated by the filler network for filler-filler and filler-polymer interaction.34 The lower Payne effect usually implies a stronger filler-polymer interaction and a better dispersion of the filler. In other words, with the increasing of TEOS from 20 to 60 phr in SBR matrix, the interaction between silica-silica was gradually increased. This meant the dispersion of silica in the SBR matrix was decreased and directly led to the increasing of G’. For the case of neat SBR, due to the absence of silica in the SBR matrix, the G’ for the neat SBR was lower than those containing silica in the SBR matrix.
The static mechanical properties of the neat SBR and different contents of silica in the SBR matrix are shown in Figure 8. It is well known that the tensile properties are affected by the size of agglomerates formed by the filler,35,36 the rubber matrix and the filler interaction.37 For the samples that are weak or are without rubber-filler chemical bonding, the fracture first occurs at the rubber-filler interface during the stretching.38 As can be seen in Figure 8, a weak shoulder was shown at the beginning of the silica-filled SBR matrix. This was mainly due to the strong filler-filler network in the SBR matrix. As the TEOS contents increased from 20 to 60 phr, the filler-filler interaction was clearly enhanced, and the shoulder also seemed more clearly compared with the neat SBR. Furthermore, the modulus at 100% and 300% among all the samples were increased gradually with the increasing of silica contents. The elongation at break for all the samples also exhibited the same trend, indicated a higher hardness among them This may be due to the chemical bond formation during the high temperature of the experiment process.
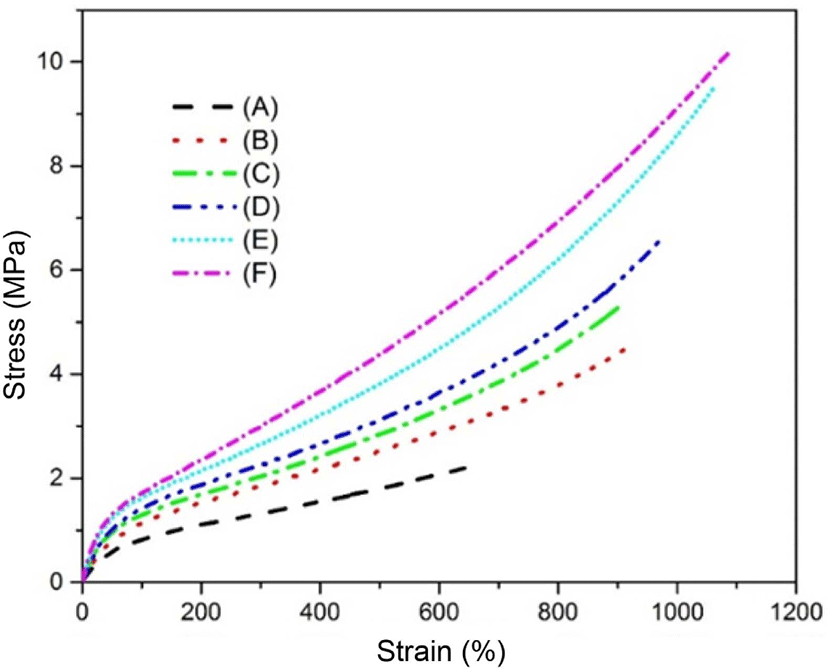
The shore A hardness of the different silica contents in the SBR matrix was stronger than that of the neat SBR, mainly due to the rigid phase of silica in the SBR matrix. The hardness of the silica-filled rubber matrix increased with the increasing of silica contents, which meant the silica particles contained a higher level of filler-filler networks with the increasing of silica contents gradually. Accordingly, the in situ silica generated by the method in this paper could act as “soft-composite materials” with relatively good mechanical properties in terms of the modulus.39
Due to the serious aggregation of silica in the SBR matrix, the silane coupling agents of TESPT, GPTS, and ETS were applied to improve the dispersion of silica in the SBR matrix. Based on the SEM graphs, 50 phr of TEOS and 8% of silane coupling agents were used in the modified system. And after the surface modification of silica with silane coupling agents, such as TESPT, GPTS, and ETS, the minimum torque for sample (E) was apparently reduced, and a decrease in the viscosity of the system was indicated after adding the silane coupling agents. This corresponds with the results reported by Choi’s group.40 Furthermore, the samples modified by silane coupling agents exhibited the higher ?M than the samples un-modified by silane coupling agents. This suggested that the silanol groups in silane coupling agents generated crosslinks between the rubber chains and increased the crosslink degree of as-prepared samples.41
For case of ts2 and t90, TESPT, GPTS, and ETS modified silica in the rubber matrix almost showed the same value as the un-modified silica sample (E) (seen in Table 2), indicated the silane coupling agent had less effect on the curing character of as-prepared samples. In other words, the cure rate index for sample (E) was nearly the same as the samples modified by silane coupling agent.
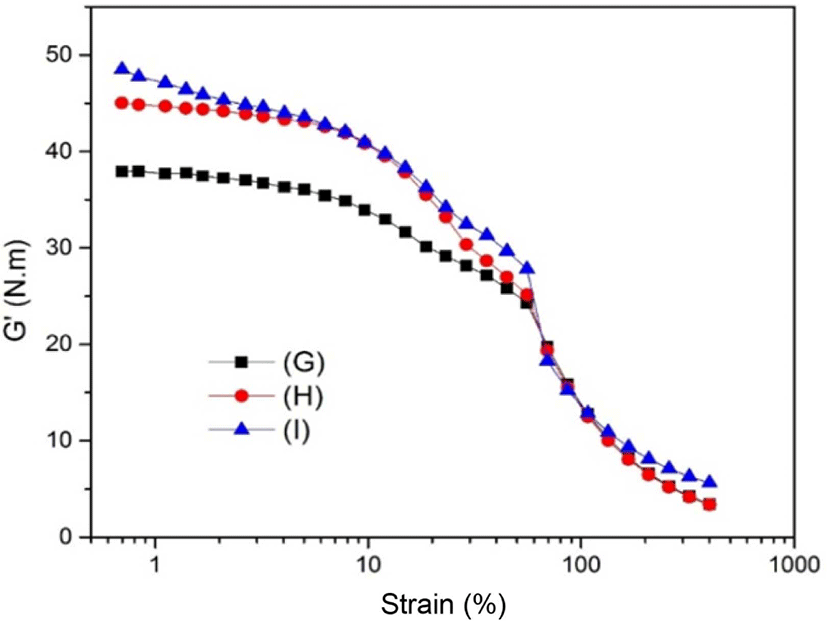
Figure 9 shows the strain sweep of as-prepared silica which modified by TESPT, GPTS, and ETS in the SBR matrix. As can be seen, the strain for the modified silica also showed the same trend as the samples without silane coupling agent modification (seen in Figure 7). The G’ of sample (G) shown in Figure 9 was lower than that of sample (H) and (I). There might be two reasons for why this occurred. First, the TESPT (as a bi-functional silane coupling agent) chemically bonded between the silica surface and the rubber matrix. This could have reduced the polarity of the silica particles and improved the dispersion of silica in the SBR matrix.42,43 The other reason was mainly due to the structure of TESPT tended to form the net structure after modification on the surface of the silica. This meant the TESPT-modified silica was easier to crosslink with the SBR matrix polymer and formed a uniform distribution in the SBR matrix (shown in Figure 10). Furthermore, each TESPT molecule contains six -OC2H5 groups, meant it could contact with more silica molecules compared with GPTS and ETS. While GPTS and ETS modified silica in the SBR matrix seemed only to be shown the long chain in the SBR matrix. They exhibited a less uniform structure and higher G’ in Figure 8. In addition, the lower G’ of sample (G) indicated an enhanced dispersion of silica. This meant the filler-filler network was reduced and the interfacial interaction between the filler and SBR matrix was improved.44,45 For the case of modified silica, the G’ of silane coupling agents modified silica in the SBR matrix was lower than that of sample (E) without silane coupling agent modification in Figure 7. This was mainly due to the improved dispersion of silica in SBR matrix after the surface of silica was modified by silane coupling agents.
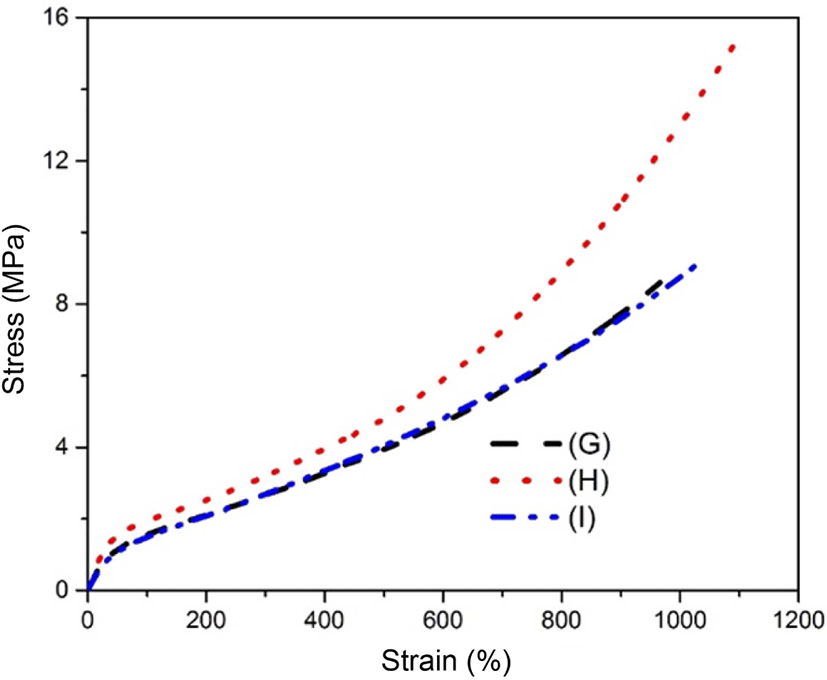
The static mechanical properties of the silane coupling agents modified silica in the SBR matrix are shown in Figure 11. As can be seen in the figure, the tensile stress and the elongation at break for sample (H) were obviously higher than those for sample (G) and (I). This could be ascribed to the lone-pair effect of the oxygen atom in GPTS come into contact with the SBR matrix. This effect directly led to the higher modulus of sample (H). Moreover, the shore A hardness for sample (H) was also stronger compared with the sample (G) and (I), also indicated a higher tensile stress of sample (H). In addition, the shore A hardness for sample (G) and (I) seemed a slightly lower than that for sample (E). This meant that there was a higher tensile stress of sample (E). For the case of sample (H), its hardness was stronger than that of sample (E). In other words, the tensile stress for sample (H) (15.34 MPa) was higher than that for sample (E) (9.48 MPa).
Conclusions
The different contents of silica in the SBR matrix were prepared based on the TEOS converted into silica sol over the acid catalyst. The mixtures were blended with the SBR latex, and the silica-reinforced SBR was subsequently obtained after dried in an oven based on the modified in situ sol-gel process. The structures of the as-prepared samples were characterized by various measurements and the mechanical properties of as-prepared samples were systematically discussed in this paper. The results in the SEM graphs exhibited the fracture surface became rather rough with the increasing of silica contents. However, the evenness of the fracture surface for the sample seemed to be improved after the silica was modified by TESPT, GPTS, and ETS silane coupling agents. XPS measurement verified the Si valence after the sample was dried in the SBR matrix. FT-IR also showed the formation of Si-O bond. Furthermore, the TGA measurement showed the decomposition temperature for as-prepared samples was effected by the contents of silica in the SBR matrix. In addition, for the mechanical property of silica without silane coupling agent modification, the storage modulus clearly increased with the increasing of silica contents. This increased Payne effect implied a stronger filler-filler interaction and a poor dispersion of the filler in the SBR matrix. The tensile stress and the hardness both increased with the increasing of silica contents in the SBR matrix, indicated the higher interaction between filler-filler was enhanced with the increasing of silica contents. For the case of modification by TESPT, GPTS, and ETS silane coupling agents, the storage modulus was decreased compared with the silica without modification by silane coupling agents, indicated an improved dispersion of modified silica in the SBR matrix. The tensile stress for the sample modified by GPTS was apparently higher than that for the sample modified by TESPT and ETS, demonstrated a special structure of the oxygen atom lone-pair effect in GPTS. This improved sol-gel method could not only shorten the preparation process, but also broaden the preparation methods for new rubber materials in the future.